New Biophysical Approaches Reveal the Dynamics and Mechanics of Type I Viral Fusion Machinery and Their Interplay with Membranes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
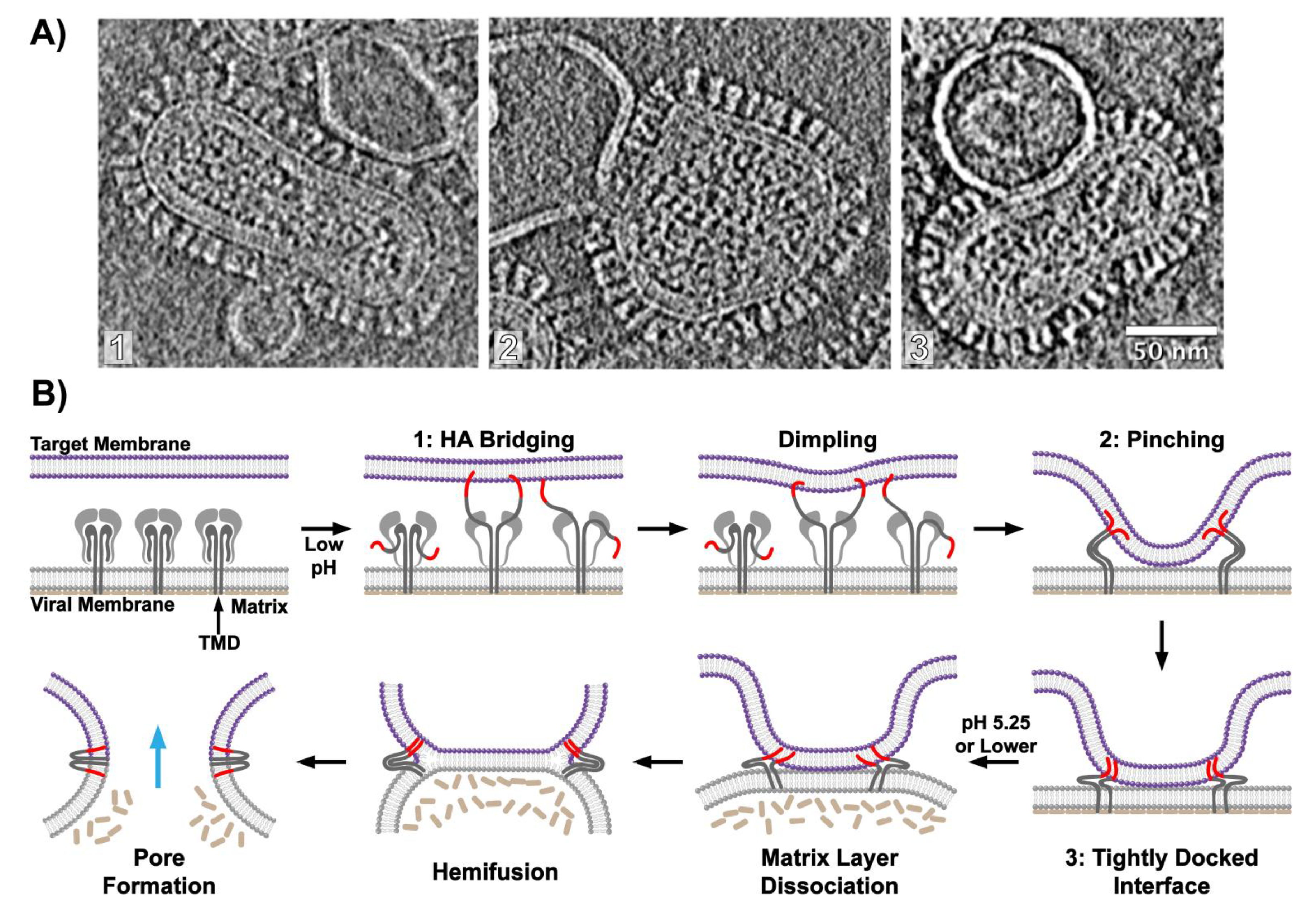
1. Introduction
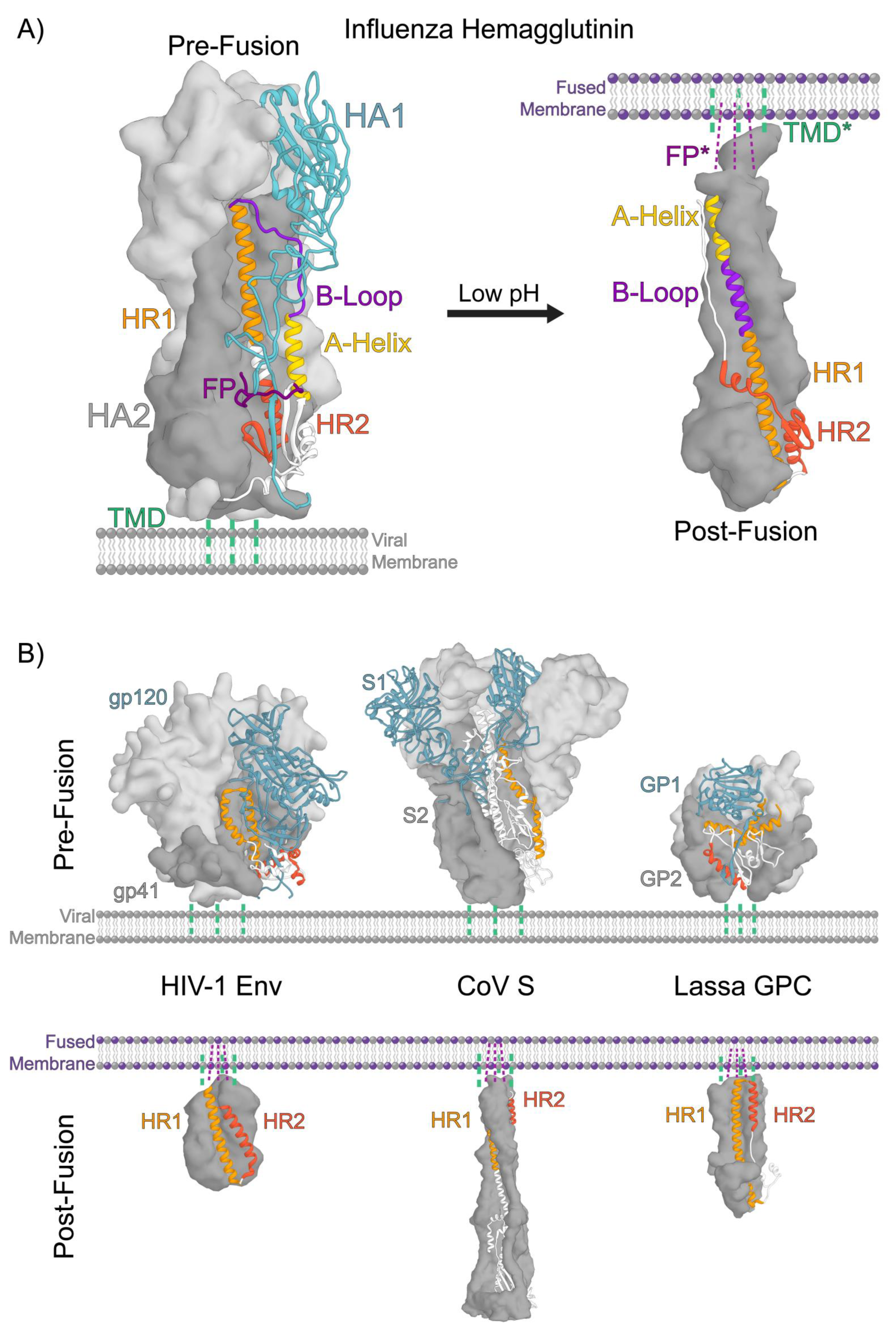
2. Structural Organization of Class 1 Viral Fusion Proteins
3. A Conserved Architecture Shared by Divergent Viruses
4. Despite their Common Architectures, Activation Mechanisms and Triggers are Highly Divergent Among Type I Fusion Proteins
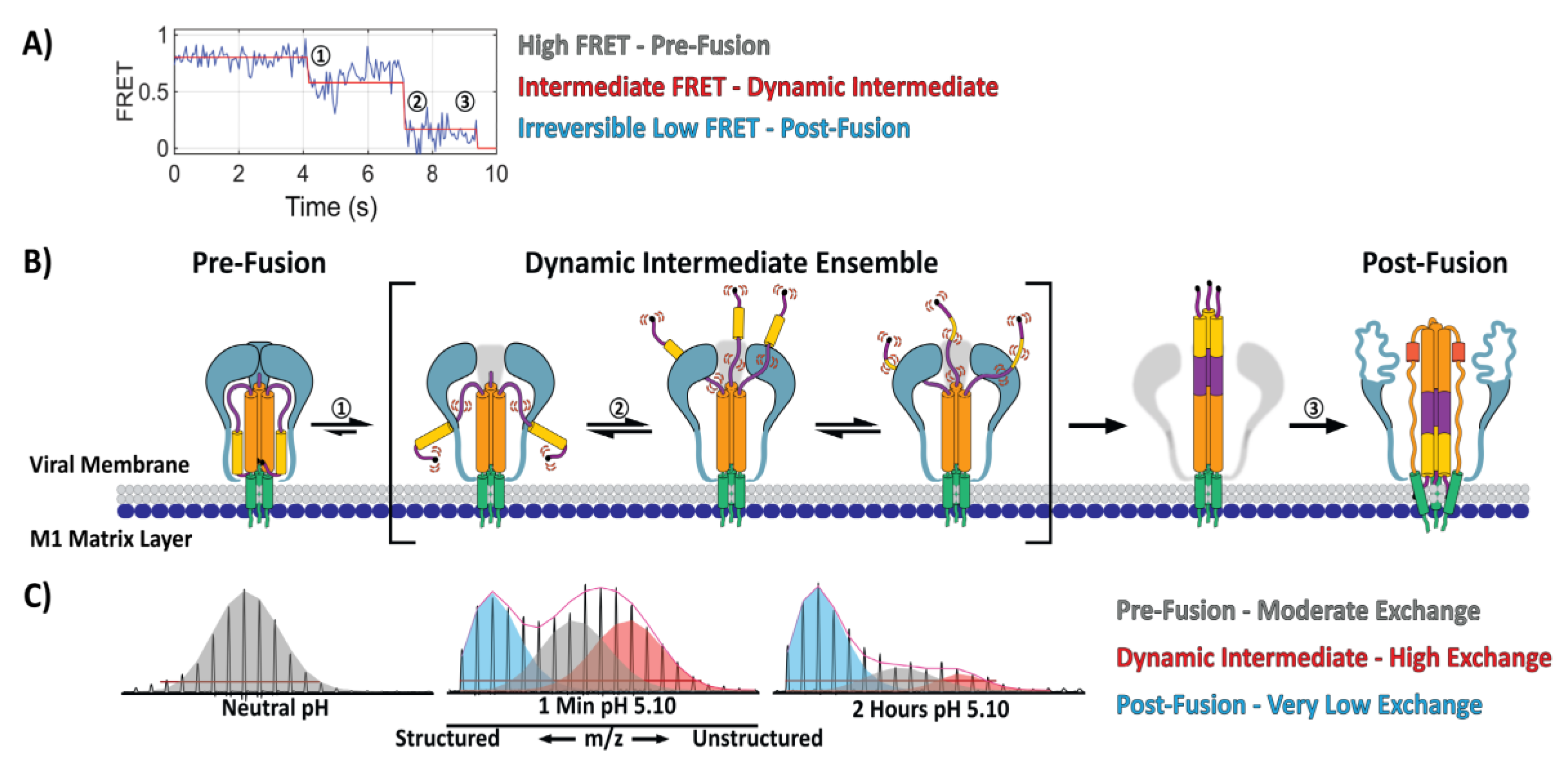
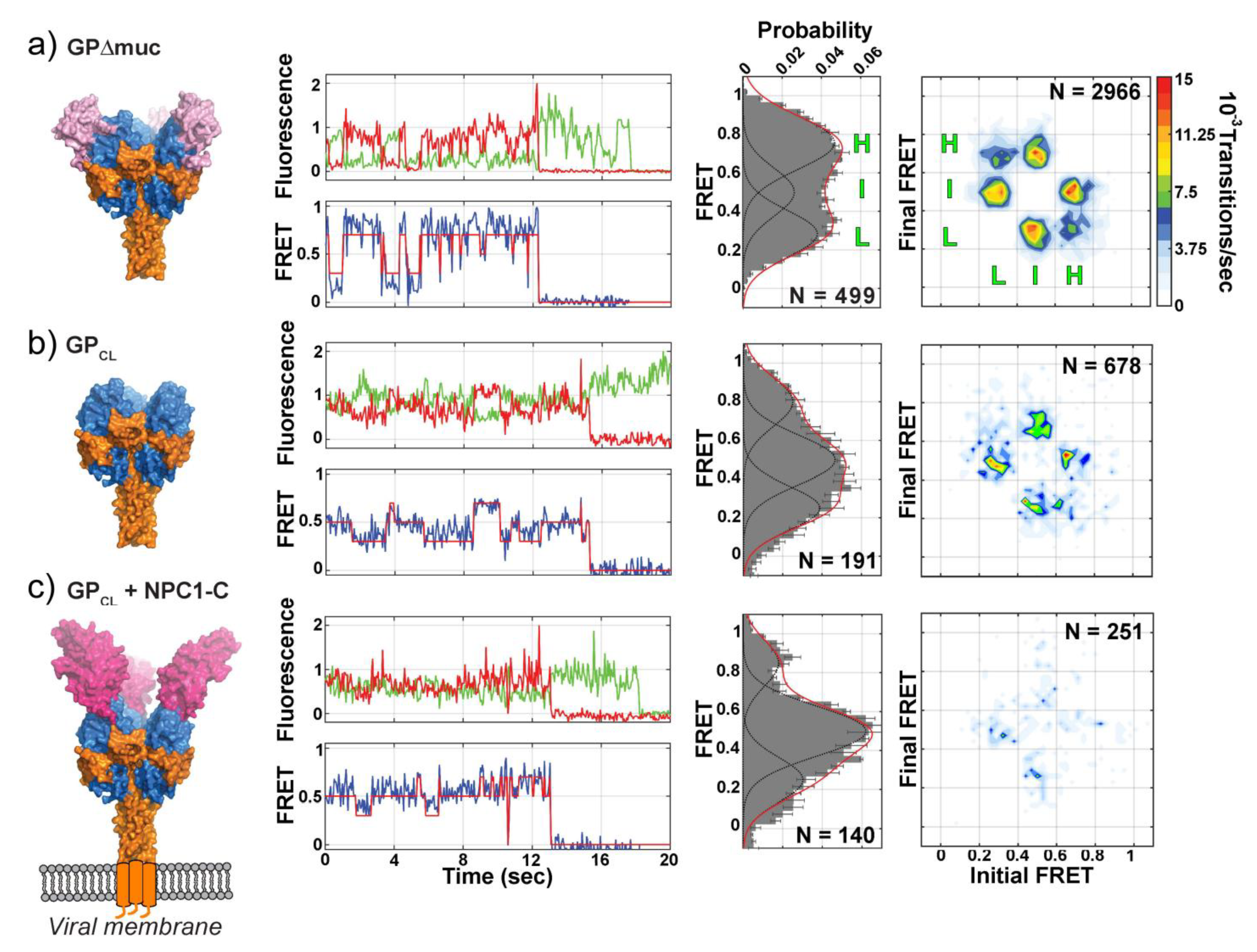
5. Direct Monitoring of the Transitions between Conformational States
6. Visualizing Viral Membrane Fusion in Action
7. Understanding HA-mediated Fusion as a First Step towards a Broader Understanding of Fusion in Diverse Enveloped Viruses
Funding
Acknowledgments
Conflicts of Interest
References
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef]
- Südhof, T.C.; Rothman, J.E. Membrane fusion: Grappling with SNARE and SM proteins. Science 2009, 323, 474–477. [Google Scholar] [CrossRef]
- McNew, J.A.; Sondermann, H.; Lee, T.; Stern, M.; Brandizzi, F. GTP-dependent membrane fusion. Annu. Rev. Cell Dev. Biol. 2013, 29, 529–550. [Google Scholar] [CrossRef]
- Rey, F.A.; Lok, S.M. Common Features of Enveloped Viruses and Implications for Immunogen Design for Next-Generation Vaccines. Cell 2018, 172, 1319–1334. [Google Scholar] [CrossRef]
- Segev, N.; Avinoam, O.; Podbilewicz, B. Fusogens. Curr. Biol. 2018, 28, R378–R380. [Google Scholar] [CrossRef]
- Jahn, R.; Lang, T.; Sudhof, T.C. Membrane fusion. Cell 2003, 112, 519–533. [Google Scholar] [CrossRef]
- Colman, P.M.; Lawrence, M.C. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell. Biol. 2003, 4, 309–319. [Google Scholar] [CrossRef]
- White, J.M.; Whittaker, G.R. Fusion of Enveloped Viruses in Endosomes. Traffic 2016, 17, 593–614. [Google Scholar] [CrossRef]
- Hernandez, J.M.; Podbilewicz, B. The hallmarks of cell-cell fusion. Development 2017, 144, 4481–4495. [Google Scholar] [CrossRef]
- Li, Y.; Modis, Y. A novel membrane fusion protein family in Flaviviridae? Trends Microbiol. 2014, 22, 176–182. [Google Scholar] [CrossRef]
- Modis, Y. Relating structure to evolution in class II viral membrane fusion proteins. Curr. Opin. Virol. 2014, 5, 34–41. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507. [Google Scholar] [CrossRef]
- Backovic, M.; Jardetzky, T.S. Class III viral membrane fusion proteins. Curr. Opin. Struct. Biol. 2009, 19, 189–196. [Google Scholar] [CrossRef]
- Kadlec, J.; Loureiro, S.; Abrescia, N.G.; Stuart, D.I.; Jones, I.M. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 2008, 15, 1024–1030. [Google Scholar] [CrossRef]
- Roche, S.; Bressanelli, S.; Rey, F.A.; Gaudin, Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 2006, 313, 187–191. [Google Scholar] [CrossRef]
- Backovic, M.; Longnecker, R.; Jardetzky, T.S. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. USA 2009, 106, 2880–2885. [Google Scholar] [CrossRef]
- Wharton, S.A.; Ruigrok, R.W.; Martin, S.R.; Skehel, J.J.; Bayley, P.M.; Weis, W.; Wiley, D.C. Conformational aspects of the acid-induced fusion mechanism of influenza virus hemagglutinin. Circular dichroism and fluorescence studies. J. Biol. Chem. 1988, 263, 4474–4480. [Google Scholar]
- Korte, T.; Herrmann, A. pH-dependent binding of the fluorophore bis-ANS to influenza virus reflects the conformational change of hemagglutinin. Eur. Biophys. J. 1994, 23, 105–113. [Google Scholar] [CrossRef]
- Korte, T.; Ludwig, K.; Krumbiegel, M.; Zirwer, D.; Damaschun, G.; Herrmann, A. Transient changes of the conformation of hemagglutinin of influenza virus at low pH detected by time-resolved circular dichroism spectroscopy. J. Biol. Chem. 1997, 272, 9764–9770. [Google Scholar] [CrossRef]
- Korte, T.; Ludwig, K.; Booy, F.P.; Blumenthal, R.; Herrmann, A. Conformational intermediates and fusion activity of influenza virus hemagglutinin. J. Virol. 1999, 73, 4567–4574. [Google Scholar] [CrossRef]
- Carr, C.M.; Chaudhry, C.; Kim, P.S. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. USA 1997, 94, 14306–14313. [Google Scholar] [CrossRef]
- Das, D.K.; Govindan, R.; Nikic-Spiegel, I.; Krammer, F.; Lemke, E.A.; Munro, J.B. Direct Visualization of the Conformational Dynamics of Single Influenza Hemagglutinin Trimers. Cell 2018, 174, 926–937.e12. [Google Scholar] [CrossRef]
- Klenk, H.D.; Rott, R.; Orlich, M.; Blodorn, J. Activation of influenza A viruses by trypsin treatment. Virology 1975, 68, 426–439. [Google Scholar] [CrossRef]
- Wilson, I.A.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 1981, 289, 366–373. [Google Scholar] [CrossRef]
- Skehel, J.J.; Bayley, P.M.; Brown, E.B.; Martin, S.R.; Waterfield, M.D.; White, J.M.; Wilson, I.A.; Wiley, D.C. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus-mediated membrane fusion. Proc. Natl. Acad. Sci. USA 1982, 79, 968–972. [Google Scholar] [CrossRef]
- White, J.; Kartenbeck, J.; Helenius, A. Membrane fusion activity of influenza virus. EMBO J. 1982, 1, 217–222. [Google Scholar] [CrossRef]
- White, J.M.; Wilson, I.A. Anti-peptide antibodies detect steps in a protein conformational change: Low-pH activation of the influenza virus hemagglutinin. J. Cell Biol. 1987, 105, 2887–2896. [Google Scholar] [CrossRef]
- Godley, L.; Pfeifer, J.; Steinhauer, D.; Ely, B.; Shaw, G.; Kaufmann, R.; Suchanek, E.; Pabo, C.; Skehel, J.J.; Wiley, D.C.; et al. Introduction of intersubunit disulfide bonds in the membrane-distal region of the influenza hemagglutinin abolishes membrane fusion activity. Cell 1992, 68, 635–645. [Google Scholar] [CrossRef]
- Kemble, G.W.; Bodian, D.L.; Rosé, J.; Wilson, I.A.; White, J.M. Intermonomer disulfide bonds impair the fusion activity of influenza virus hemagglutinin. J. Virol. 1992, 66, 4940–4950. [Google Scholar] [CrossRef]
- Carr, C.M.; Kim, P.S. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 1993, 73, 823–832. [Google Scholar] [CrossRef]
- Chen, J.; Skehel, J.J.; Wiley, D.C. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA2 subunit to form an N cap that terminates the triple-stranded coiled coil. Proc. Natl. Acad. Sci. USA 1999, 96, 8967–8972. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, S.; Blijleven, J.S.; Roos, W.H.; Onck, P.R.; van der Giessen, E.; van Oijen, A.M. Hemagglutinin-Mediated Membrane Fusion: A Biophysical Perspective. Annu. Rev. Biophys. 2018, 47, 153–173. [Google Scholar] [CrossRef] [PubMed]
- Francois-Martin, C.; Rothman, J.E.; Pincet, F. Low energy cost for optimal speed and control of membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 1238–1241. [Google Scholar] [CrossRef]
- Monck, J.R.; Fernandez, J.M. The exocytotic fusion pore. J. Cell Biol. 1992, 119, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Chanturiya, A.; Chernomordik, L.V.; Zimmerberg, J. Flickering fusion pores comparable with initial exocytotic pores occur in protein-free phospholipid bilayers. Proc. Natl. Acad. Sci. USA 1997, 94, 14423–14428. [Google Scholar] [CrossRef]
- Chernomordik, L.V.; Kozlov, M.M. Mechanics of membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 675–683. [Google Scholar] [CrossRef]
- Lorieau, J.L.; Louis, J.M.; Bax, A. The complete influenza hemagglutinin fusion domain adopts a tight helical hairpin arrangement at the lipid:water interface. Proc. Natl. Acad. Sci. USA 2010, 107, 11341–11346. [Google Scholar] [CrossRef]
- Nanavati, C.; Markin, V.S.; Oberhauser, A.F.; Fernandez, J.M. The exocytotic fusion pore modeled as a lipidic pore. Biophys. J. 1992, 63, 1118–1132. [Google Scholar] [CrossRef]
- Aeffner, S.; Reusch, T.; Weinhausen, B.; Salditt, T. Energetics of stalk intermediates in membrane fusion are controlled by lipid composition. Proc. Natl. Acad. Sci. USA 2012, 109, E1609–E1618. [Google Scholar] [CrossRef]
- Kemble, G.W.; Danieli, T.; White, J.M. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell 1994, 76, 383–391. [Google Scholar] [CrossRef]
- Blumenthal, R.; Sarkar, D.P.; Durell, S.; Howard, D.E.; Morris, S.J. Dilation of the influenza hemagglutinin fusion pore revealed by the kinetics of individual cell-cell fusion events. J. Cell Biol. 1996, 135, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.C.; McMullan, G.; Scheres, S.H. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 2015, 40, 49–57. [Google Scholar] [CrossRef]
- Hryc, C.F.; Chen, D.H.; Chiu, W. Near-atomic-resolution cryo-EM for molecular virology. Curr. Opin. Virol. 2011, 1, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Garcia, N.K.; Lee, K.K. Dynamic Viral Glycoprotein Machines: Approaches for Probing Transient States That Drive Membrane Fusion. Viruses 2016, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.G. Structure of viruses: A short history. Q. Rev. Biophys. 2013, 46, 133–180. [Google Scholar] [CrossRef]
- Stass, R.; Ilca, S.L.; Huiskonen, J.T. Beyond structures of highly symmetric purified viral capsids by cryo-EM. Curr. Opin. Struct. Biol. 2018, 52, 25–31. [Google Scholar] [CrossRef]
- Fontana, J.; Steven, A.C. Influenza virus-mediated membrane fusion: Structural insights from electron microscopy. Arch. Biochem. Biophys. 2015, 581, 86–97. [Google Scholar] [CrossRef]
- Zhou, Z.H. Structures of viral membrane proteins by high-resolution cryoEM. Curr. Opin. Virol. 2014, 5, 111–119. [Google Scholar] [CrossRef]
- Guttman, M.; Lee, K.K. Isotope Labeling of Biomolecules: Structural Analysis of Viruses by HDX-MS. Methods Enzymol. 2016, 566, 405–426. [Google Scholar] [CrossRef]
- Tian, Z.; Gong, J.; Crowe, M.; Lei, M.; Li, D.; Ji, B.; Diao, J. Biochemical studies of membrane fusion at the single-particle level. Prog. Lipid. Res. 2019, 73, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Otterstrom, J.; van Oijen, A.M. Visualization of membrane fusion, one particle at a time. Biochemistry 2013, 52, 1654–1668. [Google Scholar] [CrossRef] [PubMed]
- Caston, J.R. Conventional electron microscopy, cryo-electron microscopy and cryo-electron tomography of viruses. Subcell. Biochem. 2013, 68, 79–115. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Liu, X.; Rochat, R.H.; Baker, M.L.; Chiu, W. Reconstructing virus structures from nanometer to near-atomic resolutions with cryo-electron microscopy and tomography. Adv. Exp. Med. Biol. 2012, 726, 49–90. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A. Structural biology in situ—The potential of subtomogram averaging. Curr. Opin. Struct. Biol. 2013, 23, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Benhaim, M.; Mangala Prasad, V.; Garcia, N.K.; Guttman, M.; Lee, K.K. Structural monitoring of a transient intermediate in the hemagglutinin fusion machinery on influenza virions. Sci. Adv. 2020, in press. [Google Scholar]
- Lazarowitz, S.G.; Choppin, P.W. Enhancement of the infectivity of influenza A and B viruses by proteolytic cleavage of the hemagglutinin polypeptide. Virology 1975, 68, 440–454. [Google Scholar] [CrossRef]
- Chen, J.; Lee, K.H.; Steinhauer, D.A.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998, 95, 409–417. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Veesler, D. Structural insights into coronavirus entry. Adv. Virus. Res. 2019, 105, 93–116. [Google Scholar] [CrossRef]
- Limburg, H.; Harbig, A.; Bestle, D.; Stein, D.A.; Moulton, H.M.; Jaeger, J.; Janga, H.; Hardes, K.; Koepke, J.; Schulte, L.; et al. TMPRSS2 Is the Major Activating Protease of Influenza A Virus in Primary Human Airway Cells and Influenza B Virus in Human Type II Pneumocytes. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Klenk, H.D.; Rott, R. The molecular biology of influenza virus pathogenicity. Adv. Virus Res. 1988, 34, 247–281. [Google Scholar] [CrossRef] [PubMed]
- Vey, M.; Orlich, M.; Adler, S.; Klenk, H.D.; Rott, R.; Garten, W. Hemagglutinin activation of pathogenic avian influenza viruses of serotype H7 requires the protease recognition motif R-X-K/R-R. Virology 1992, 188, 408–413. [Google Scholar] [CrossRef]
- Klenk, H.D.; Garten, W. Host cell proteases controlling virus pathogenicity. Trends Microbiol. 1994, 2, 39–43. [Google Scholar] [CrossRef]
- Bullough, P.A.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994, 371, 37–43. [Google Scholar] [CrossRef]
- Benton, D.J.; Nans, A.; Calder, L.J.; Turner, J.; Neu, U.; Lin, Y.P.; Ketelaars, E.; Kallewaard, N.L.; Corti, D.; Lanzavecchia, A.; et al. Influenza hemagglutinin membrane anchor. Proc. Natl. Acad. Sci. USA 2018, 115, 10112–10117. [Google Scholar] [CrossRef]
- Park, H.E.; Gruenke, J.A.; White, J.M. Leash in the groove mechanism of membrane fusion. Nat. Struct. Biol. 2003, 10, 1048–1053. [Google Scholar] [CrossRef]
- Welch, B.D.; Liu, Y.; Kors, C.A.; Leser, G.P.; Jardetzky, T.S.; Lamb, R.A. Structure of the cleavage-activated prefusion form of the parainfluenza virus 5 fusion protein. Proc. Natl. Acad. Sci. USA 2012, 109, 16672–16677. [Google Scholar] [CrossRef]
- Hastie, K.M.; Zandonatti, M.A.; Kleinfelter, L.M.; Heinrich, M.L.; Rowland, M.M.; Chandran, K.; Branco, L.M.; Robinson, J.E.; Garry, R.F.; Saphire, E.O. Structural basis for antibody-mediated neutralization of Lassa virus. Science 2017, 356, 923–928. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Bosch, B.J.; Frenz, B.; Rottier, P.J.M.; DiMaio, F.; Rey, F.A.; Veesler, D. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 2016, 531, 114–117. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef]
- Swanson, K.A.; Settembre, E.C.; Shaw, C.A.; Dey, A.K.; Rappuoli, R.; Mandl, C.W.; Dormitzer, P.R.; Carfi, A. Structural basis for immunization with postfusion respiratory syncytial virus fusion F glycoprotein (RSV F) to elicit high neutralizing antibody titers. Proc. Natl. Acad. Sci. USA 2011, 108, 9619–9624. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Yang, Y.; Graham, B.S.; Kwong, P.D. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J. Virol. 2011, 85, 7788–7796. [Google Scholar] [CrossRef] [PubMed]
- Malashkevich, V.N.; Schneider, B.J.; McNally, M.L.; Milhollen, M.A.; Pang, J.X.; Kim, P.S. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9-A resolution. Proc. Natl. Acad. Sci. USA 1999, 96, 2662–2667. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Fusco, M.L.; Hessell, A.J.; Oswald, W.B.; Burton, D.R.; Saphire, E.O. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 2008, 454, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Ozorowski, G.; Ward, A.B. Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science 2016, 351, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Stoller, M.O.; Wang, S.; Liu, J.; Fagan, M.B.; Nunberg, J.H. Structural and functional analysis of interhelical interactions in the human immunodeficiency virus type 1 gp41 envelope glycoprotein by alanine-scanning mutagenesis. J. Virol. 2001, 75, 11146–11156. [Google Scholar] [CrossRef]
- Chen, J.; Wharton, S.A.; Weissenhorn, W.; Calder, L.J.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in Escherichia coli into the low-pH-induced conformation. Proc. Natl. Acad. Sci. USA 1995, 92, 12205–12209. [Google Scholar] [CrossRef]
- Blijleven, J.S.; Boonstra, S.; Onck, P.R.; van der Giessen, E.; van Oijen, A.M. Mechanisms of influenza viral membrane fusion. Semin. Cell Dev. Biol. 2016, 60, 78–88. [Google Scholar] [CrossRef]
- Williams, J.A.; Gui, L.; Hom, N.; Mileant, A.; Lee, K.K. Dissection of epitope-specific mechanisms of neutralization of influenza virus by intact IgG and Fab fragments. J. Virol. 2017. [Google Scholar] [CrossRef]
- Fontana, J.; Cardone, G.; Heymann, J.B.; Winkler, D.C.; Steven, A.C. Structural changes in Influenza virus at low pH characterized by cryo-electron tomography. J. Virol. 2012, 86, 2919–2929. [Google Scholar] [CrossRef]
- Garcia, N.K.; Guttman, M.; Ebner, J.L.; Lee, K.K. Dynamic changes during acid-induced activation of influenza hemagglutinin. Structure 2015, 23, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Benhaim, M.; Lee, K.K.; Guttman, M. Tracking Higher Order Protein Structure by Hydrogen-Deuterium Exchange Mass Spectrometry. Protein Pept. Lett. 2019, 26, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Hodge, E.A.; Benhaim, M.A.; Lee, K.K. Bridging protein structure, dynamics, and function using hydrogen/deuterium-exchange mass spectrometry. Protein Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Leikina, E.; Ramos, C.; Markovic, I.; Zimmerberg, J.; Chernomordik, L.V. Reversible stages of the low-pH-triggered conformational change in influenza virus hemagglutinin. EMBO J. 2002, 21, 5701–5710. [Google Scholar] [CrossRef]
- Xu, R.; Wilson, I.A. Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J. Virol. 2011, 85, 5172–5182. [Google Scholar] [CrossRef]
- Lin, X.; Eddy, N.R.; Noel, J.K.; Whitford, P.C.; Wang, Q.; Ma, J.; Onuchic, J.N. Order and disorder control the functional rearrangement of influenza hemagglutinin. Proc. Natl. Acad. Sci. USA 2014, 111, 12049–12054. [Google Scholar] [CrossRef]
- Lin, X.; Noel, J.K.; Wang, Q.; Ma, J.; Onuchic, J.N. Lowered pH Leads to Fusion Peptide Release and a Highly Dynamic Intermediate of Influenza Hemagglutinin. J. Phys. Chem. B 2016, 120, 9654–9660. [Google Scholar] [CrossRef]
- Guttman, M.; Garcia, N.K.; Cupo, A.; Matsui, T.; Julien, J.P.; Sanders, R.W.; Wilson, I.A.; Moore, J.P.; Lee, K.K. CD4-induced activation in a soluble HIV-1 Env trimer. Structure 2014, 22, 974–984. [Google Scholar] [CrossRef]
- Ward, A.B.; Wilson, I.A. Insights into the trimeric HIV-1 envelope glycoprotein structure. Trends Biochem. Sci. 2015, 40, 101–107. [Google Scholar] [CrossRef]
- Ma, X.; Lu, M.; Gorman, J.; Terry, D.S.; Hong, X.; Zhou, Z.; Zhao, H.; Altman, R.B.; Arthos, J.; Blanchard, S.C.; et al. HIV-1 Env trimer opens through an asymmetric intermediate in which individual protomers adopt distinct conformations. Elife 2018, 7. [Google Scholar] [CrossRef]
- Torrents de la Pena, A.; Rantalainen, K.; Cottrell, C.A.; Allen, J.D.; van Gils, M.J.; Torres, J.L.; Crispin, M.; Sanders, R.W.; Ward, A.B. Similarities and differences between native HIV-1 envelope glycoprotein trimers and stabilized soluble trimer mimetics. PLoS Pathog 2019, 15, e1007920. [Google Scholar] [CrossRef] [PubMed]
- Pancera, M.; Changela, A.; Kwong, P.D. How HIV-1 entry mechanism and broadly neutralizing antibodies guide structure-based vaccine design. Curr. Opin. HIV AIDS 2017, 12, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Rojek, J.M.; Kunz, S. Cell entry by human pathogenic arenaviruses. Cell Microbiol. 2008, 10, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Jae, L.T.; Raaben, M.; Herbert, A.S.; Kuehne, A.I.; Wirchnianski, A.S.; Soh, T.K.; Stubbs, S.H.; Janssen, H.; Damme, M.; Saftig, P.; et al. Virus entry. Lassa virus entry requires a trigger-induced receptor switch. Science 2014, 344, 1506–1510. [Google Scholar] [CrossRef]
- Cao, W.; Henry, M.D.; Borrow, P.; Yamada, H.; Elder, J.H.; Ravkov, E.V.; Nichol, S.T.; Compans, R.W.; Campbell, K.P.; Oldstone, M.B. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 1998, 282, 2079–2081. [Google Scholar] [CrossRef]
- Hastie, K.M.; Saphire, E.O. Lassa virus glycoprotein: Stopping a moving target. Curr. Opin. Virol. 2018, 31, 52–58. [Google Scholar] [CrossRef]
- Cohen-Dvashi, H.; Cohen, N.; Israeli, H.; Diskin, R. Molecular Mechanism for LAMP1 Recognition by Lassa Virus. J. Virol. 2015, 89, 7584–7592. [Google Scholar] [CrossRef]
- Cohen-Dvashi, H.; Israeli, H.; Shani, O.; Katz, A.; Diskin, R. Role of LAMP1 Binding and pH Sensing by the Spike Complex of Lassa Virus. J. Virol. 2016, 90, 10329–10338. [Google Scholar] [CrossRef]
- Li, S.; Sun, Z.; Pryce, R.; Parsy, M.L.; Fehling, S.K.; Schlie, K.; Siebert, C.A.; Garten, W.; Bowden, T.A.; Strecker, T.; et al. Acidic pH-Induced Conformations and LAMP1 Binding of the Lassa Virus Glycoprotein Spike. PLoS Pathog 2016, 12, e1005418. [Google Scholar] [CrossRef]
- Hulseberg, C.E.; Fénéant, L.; Szymańska, K.M.; White, J.M. Lamp1 Increases the Efficiency of Lassa Virus Infection by Promoting Fusion in Less Acidic Endosomal Compartments. mBio 2018, 9. [Google Scholar] [CrossRef]
- Porotto, M.; Salah, Z.W.; Gui, L.; DeVito, I.; Jurgens, E.M.; Lu, H.; Yokoyama, C.C.; Palermo, L.M.; Lee, K.K.; Moscona, A. Regulation of paramyxovirus fusion activation: The hemagglutinin-neuraminidase protein stabilizes the fusion protein in a pretriggered state. J. Virol. 2012, 86, 12838–12848. [Google Scholar] [CrossRef] [PubMed]
- Jardetzky, T.S.; Lamb, R.A. Activation of paramyxovirus membrane fusion and virus entry. Curr. Opin. Virol. 2014, 5, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Noel, J.K.; Wang, Q.; Ma, J.; Onuchic, J.N. Atomistic simulations indicate the functional loop-to-coiled-coil transition in influenza hemagglutinin is not downhill. Proc. Natl. Acad. Sci. USA 2018, 115, E7905–E7913. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K.; Bulow, U.; Diehl, W.E.; Durham, N.D.; Senjobe, F.; Chandran, K.; Luban, J.; Munro, J.B. Conformational changes in the Ebola virus membrane fusion machine induced by pH, Ca2+, and receptor binding. PLoS Biol 2020, 18, e3000626. [Google Scholar] [CrossRef] [PubMed]
- Durham, N.D.; Howard, A.R.; Govindan, R.; Senjobe, F.; Fels, J.M.; Diehl, W.E.; Luban, J.; Chandran, K.; Munro, J.B. Real-Time Analysis of Individual Ebola Virus Glycoproteins Reveals Pre-Fusion, Entry-Relevant Conformational Dynamics. Viruses 2020, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Saphire, E.O. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009, 4, 621–635. [Google Scholar] [CrossRef]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 2005, 308, 1643–1645. [Google Scholar] [CrossRef]
- Schornberg, K.; Matsuyama, S.; Kabsch, K.; Delos, S.; Bouton, A.; White, J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006, 80, 4174–4178. [Google Scholar] [CrossRef]
- Nanbo, A.; Imai, M.; Watanabe, S.; Noda, T.; Takahashi, K.; Neumann, G.; Halfmann, P.; Kawaoka, Y. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog 2010, 6, e1001121. [Google Scholar] [CrossRef]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef]
- Simmons, J.A.; D’Souza, R.S.; Ruas, M.; Galione, A.; Casanova, J.E.; White, J.M. Ebolavirus Glycoprotein Directs Fusion through NPC1+ Endolysosomes. J. Virol. 2016, 90, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Nathan, L.; Lai, A.L.; Millet, J.K.; Straus, M.R.; Freed, J.H.; Whittaker, G.R.; Daniel, S. Calcium Ions Directly Interact with the Ebola Virus Fusion Peptide To Promote Structure-Function Changes That Enhance Infection. ACS Infect. Dis. 2020, 6, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K. Architecture of a nascent viral fusion pore. EMBO J. 2010, 29, 1299–1311. [Google Scholar] [CrossRef]
- Chlanda, P.; Mekhedov, E.; Waters, H.; Schwartz, C.L.; Fischer, E.R.; Ryham, R.J.; Cohen, F.S.; Blank, P.S.; Zimmerberg, J. The hemifusion structure induced by influenza virus haemagglutinin is determined by physical properties of the target membranes. Nat. Microbiol. 2016, 1, 16050. [Google Scholar] [CrossRef]
- Gui, L.; Ebner, J.L.; Mileant, A.; Williams, J.A.; Lee, K.K. Visualization and Sequencing of Membrane Remodeling Leading to Influenza Virus Fusion. J. Virol. 2016, 90, 6948–6962. [Google Scholar] [CrossRef] [PubMed]
- Chernomordik, L.V.; Kozlov, M.M. Membrane hemifusion: Crossing a chasm in two leaps. Cell 2005, 123, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Yin, S.R.; Blank, P.S.; Zimmerberg, J. Cholesterol promotes hemifusion and pore widening in membrane fusion induced by influenza hemagglutinin. J. Gen. Physiol. 2008, 131, 503–513. [Google Scholar] [CrossRef]
- Risselada, H.J.; Bubnis, G.; Grubmuller, H. Expansion of the fusion stalk and its implication for biological membrane fusion. Proc. Natl. Acad. Sci. USA 2014, 111, 11043–11048. [Google Scholar] [CrossRef]
- Grunewald, K. Viral fusion: How Flu induces dimples on liposomes. EMBO J. 2010, 29, 1165–1166. [Google Scholar] [CrossRef]
- Calder, L.J.; Rosenthal, P.B. Cryomicroscopy provides structural snapshots of influenza virus membrane fusion. Nat. Struct. Mol. Biol. 2016, 23, 853–858. [Google Scholar] [CrossRef]
- Chernomordik, L.V.; Kozlov, M.M. Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 2003, 72, 175–207. [Google Scholar] [CrossRef] [PubMed]
- Efrat, A.; Chernomordik, L.V.; Kozlov, M.M. Point-like protrusion as a prestalk intermediate in membrane fusion pathway. Biophys. J. 2007, 92, L61–L63. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, P.I.; Zimmerberg, J.; Chizmadzhev, Y.A.; Cohen, F.S. A quantitative model for membrane fusion based on low-energy intermediates. Proc. Natl. Acad. Sci. USA 2001, 98, 7235–7240. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, M.M.; Chernomordik, L.V. A mechanism of protein-mediated fusion: Coupling between refolding of the influenza hemagglutinin and lipid rearrangements. Biophys. J. 1998, 75, 1384–1396. [Google Scholar] [CrossRef]
- Maurer, U.E.; Sodeik, B.; Grunewald, K. Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry. Proc. Natl. Acad. Sci. USA 2008, 105, 10559–10564. [Google Scholar] [CrossRef]
- Harris, A.; Cardone, G.; Winkler, D.C.; Heymann, J.B.; Brecher, M.; White, J.M.; Steven, A.C. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci. USA 2006, 103, 19123–19127. [Google Scholar] [CrossRef]
- Clague, M.J.; Schoch, C.; Blumenthal, R. Delay time for influenza virus hemagglutinin-induced membrane fusion depends on hemagglutinin surface density. J. Virol. 1991, 65, 2402–2407. [Google Scholar] [CrossRef]
- Danieli, T.; Pelletier, S.L.; Henis, Y.I.; White, J.M. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J. Cell Biol. 1996, 133, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Bentz, J. Minimal aggregate size and minimal fusion unit for the first fusion pore of influenza hemagglutinin-mediated membrane fusion. Biophys. J. 2000, 78, 227–245. [Google Scholar] [CrossRef]
- Günther-Ausborn, S.; Schoen, P.; Bartoldus, I.; Wilschut, J.; Stegmann, T. Role of hemagglutinin surface density in the initial stages of influenza virus fusion: Lack of evidence for cooperativity. J. Virol. 2000, 74, 2714–2720. [Google Scholar] [CrossRef]
- Mittal, A.; Shangguan, T.; Bentz, J. Measuring pKa of activation and pKi of inactivation for influenza hemagglutinin from kinetics of membrane fusion of virions and of HA expressing cells. Biophys. J. 2002, 83, 2652–2666. [Google Scholar] [CrossRef]
- Ellens, H.; Bentz, J.; Mason, D.; Zhang, F.; White, J.M. Fusion of influenza hemagglutinin-expressing fibroblasts with glycophorin-bearing liposomes: Role of hemagglutinin surface density. Biochemistry 1990, 29, 9697–9707. [Google Scholar] [CrossRef]
- Diao, J.; Grob, P.; Cipriano, D.J.; Kyoung, M.; Zhang, Y.; Shah, S.; Nguyen, A.; Padolina, M.; Srivastava, A.; Vrljic, M.; et al. Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. Elife 2012, 1, e00109. [Google Scholar] [CrossRef]
- Hernandez, J.M.; Stein, A.; Behrmann, E.; Riedel, D.; Cypionka, A.; Farsi, Z.; Walla, P.J.; Raunser, S.; Jahn, R. Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science 2012, 336, 1581–1584. [Google Scholar] [CrossRef]
- Christensen, S.M.; Mortensen, M.W.; Stamou, D.G. Single vesicle assaying of SNARE-synaptotagmin-driven fusion reveals fast and slow modes of both docking and fusion and intrasample heterogeneity. Biophys. J. 2011, 100, 957–967. [Google Scholar] [CrossRef]
- McDargh, Z.A.; Polley, A.; O’Shaughnessy, B. SNARE-mediated membrane fusion is a two-stage process driven by entropic forces. FEBS Lett. 2018, 592, 3504–3515. [Google Scholar] [CrossRef] [PubMed]
- Imig, C.; Min, S.W.; Krinner, S.; Arancillo, M.; Rosenmund, C.; Sudhof, T.C.; Rhee, J.; Brose, N.; Cooper, B.H. The morphological and molecular nature of synaptic vesicle priming at presynaptic active zones. Neuron 2014, 84, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Tamm, L.K.; Crane, J.; Kiessling, V. Membrane fusion: A structural perspective on the interplay of lipids and proteins. Curr. Opin. Struct. Biol. 2003, 13, 453–466. [Google Scholar] [CrossRef]
- Légaré, S.; Lagüe, P. The influenza fusion peptide promotes lipid polar head intrusion through hydrogen bonding with phosphates and N-terminal membrane insertion depth. Proteins 2014, 82, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, S.; Feng, Y.; Nebioglu, F.; Heilig, R.; Picotti, P.; Helenius, A. Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza A virus cores after penetration. J. Virol. 2014, 88, 13029–13046. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Avalos, R.T.; Ponimaskin, E.; Nayak, D.P. Influenza virus assembly: Effect of influenza virus glycoproteins on the membrane association of M1 protein. J. Virol. 2000, 74, 8709–8719. [Google Scholar] [CrossRef]
- Ruigrok, R.W.; Barge, A.; Durrer, P.; Brunner, J.; Ma, K.; Whittaker, G.R. Membrane interaction of influenza virus M1 protein. Virology 2000, 267, 289–298. [Google Scholar] [CrossRef]
- Lakadamyali, M.; Rust, M.J.; Babcock, H.P.; Zhuang, X. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. USA 2003, 100, 9280–9285. [Google Scholar] [CrossRef]
- Costello, D.A.; Whittaker, G.R.; Daniel, S. Variations in pH sensitivity, acid stability, and fusogenicity of three influenza virus H3 subtypes. J. Virol. 2015, 89, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Calder, L.J.; Wasilewski, S.; Berriman, J.A.; Rosenthal, P.B. Structural organization of a filamentous influenza A virus. Proc. Natl. Acad. Sci. USA 2010, 107, 10685–10690. [Google Scholar] [CrossRef] [PubMed]
- Rossman, J.S.; Leser, G.P.; Lamb, R.A. Filamentous influenza virus enters cells via macropinocytosis. J. Virol. 2012, 86, 10950–10960. [Google Scholar] [CrossRef] [PubMed]
- Badham, M.D.; Rossman, J.S. Filamentous Influenza Viruses. Curr. Clin. Microbiol. Rep. 2016, 3, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Kielian, M. Mechanisms of Virus Membrane Fusion Proteins. Annu. Rev. Virol. 2014, 1, 171–189. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benhaim, M.A.; Lee, K.K. New Biophysical Approaches Reveal the Dynamics and Mechanics of Type I Viral Fusion Machinery and Their Interplay with Membranes. Viruses 2020, 12, 413. https://doi.org/10.3390/v12040413
Benhaim MA, Lee KK. New Biophysical Approaches Reveal the Dynamics and Mechanics of Type I Viral Fusion Machinery and Their Interplay with Membranes. Viruses. 2020; 12(4):413. https://doi.org/10.3390/v12040413
Chicago/Turabian StyleBenhaim, Mark A., and Kelly K. Lee. 2020. "New Biophysical Approaches Reveal the Dynamics and Mechanics of Type I Viral Fusion Machinery and Their Interplay with Membranes" Viruses 12, no. 4: 413. https://doi.org/10.3390/v12040413
APA StyleBenhaim, M. A., & Lee, K. K. (2020). New Biophysical Approaches Reveal the Dynamics and Mechanics of Type I Viral Fusion Machinery and Their Interplay with Membranes. Viruses, 12(4), 413. https://doi.org/10.3390/v12040413