The Potential of Long-Acting, Tissue-Targeted Synthetic Nanotherapy for Delivery of Antiviral Therapy Against HIV Infection


Abstract
1. Introduction
2. State of the ART
3. Bioavailability and Pharmacokinetics
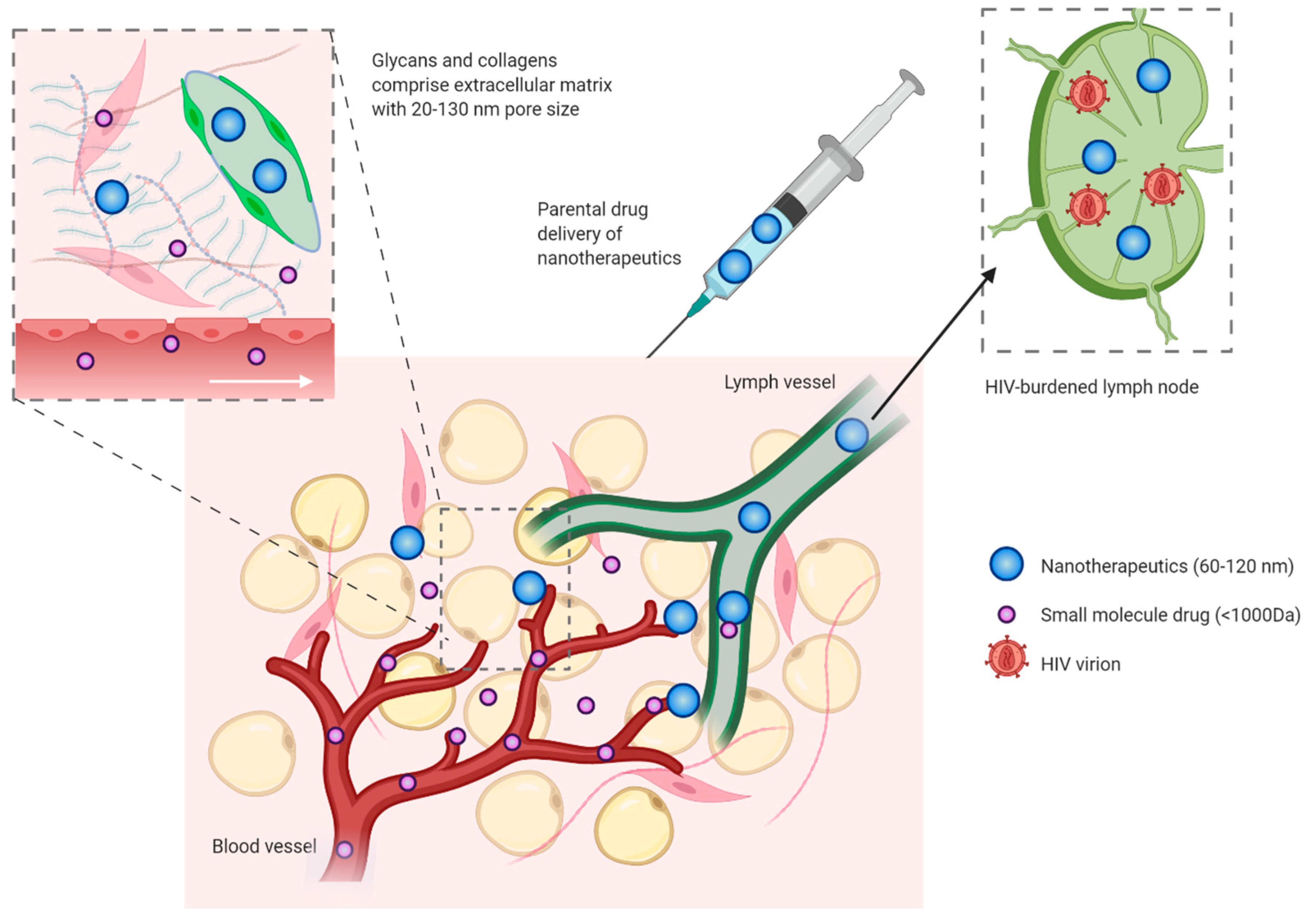
4. Tissue Penetration
5. What are Nanotherapeutics for HIV Treatment and Cure?
6. Long-acting Antiviral Prodrugs
7. Increasing Access of Drugs to Lymphoid Tissues
8. Cellular Targeting
9. Nanotherapeutics in an HIV-Cure Context
10. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- UNAIDS. Global HIV Statistics-Fact Sheet; UNAIDS 2019. Available online: https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf (accessed on 13 January 2020).
- Blankson, J.N.; Persaud, D.; Siliciano, R.F. The Challenge of Viral Reservoirs in HIV-1 Infection. Annu. Rev. Med. 2002, 53, 557–593. [Google Scholar] [CrossRef] [PubMed]
- Estes, J.D.; Kityo, C.; Ssali, F.; Swainson, L.; Makamdop, K.N.; Del Prete, G.Q.; Deeks, S.G.; Luciw, P.A.; Chipman, J.G.; Beilman, G.J.; et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat. Med. 2017, 23, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.V.; Staskus, K.; Wietgrefe, S.W.; Rothenberger, M.; Reilly, C.; Chipman, J.G.; Beilman, G.J.; Khoruts, A.; Thorkelson, A.; Schmidt, T.E.; et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Henrich, T.J.; Hatano, H.; Bacon, O.; Hogan, L.E.; Rutishauser, R.; Hill, A.; Kearney, M.F.; Anderson, E.M.; Buchbinder, S.P.; Cohen, S.E.; et al. HIV-1 persistence following extremely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: An observational study. PLoS Med. 2017, 14, e1002417. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Redondo, R.; Fryer, H.R.; Bedford, T.; Kim, E.Y.; Archer, J.; Kosakovsky Pond, S.L.; Chung, Y.S.; Penugonda, S.; Chipman, J.G.; Fletcher, C.V.; et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 2016, 530, 51–56. [Google Scholar] [CrossRef]
- Deeks, S.G. Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef]
- Berg, S.L.; Stone, J.; Xiao, J.J.; Chan, K.K.; Nuchtern, J.; Dauser, R.; McGuffey, L.; Thompson, P.; Blaney, S.M. Plasma and cerebrospinal fluid pharmacokinetics of depsipeptide (FR901228) in nonhuman primates. Cancer Chemother. Pharmacol. 2004, 54, 85–88. [Google Scholar] [CrossRef]
- Gupta, S.; Kesarla, R.; Omri, A. Approaches for CNS delivery of drugs–nose to brain targeting of antiretroviral agents as a potential attempt for complete elimination of major reservoir site of HIV to aid AIDS treatment. Expert Opin. Drug Deliv. 2019, 16, 287–300. [Google Scholar] [CrossRef]
- Varghese, N.M.; Senthil, V.; Saxena, S.K. Nanocarriers for brain specific delivery of anti-retro viral drugs: Challenges and achievements. J. Drug Target. 2018, 26, 195–207. [Google Scholar] [CrossRef]
- Davey, R.T.; Bhat, N.; Yoder, C.; Chun, T.W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.A.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef] [PubMed]
- Stagg, M.P.; Cretton, E.M.; Kidd, L.; Diasio, R.B.; Sommadossi, J.-P. Clinical pharmacokinetics of 3′-azido--3′--deoxythymidine (zidovudine) and catabolites with formation of a toxic catabolite, 3′-amino-3′-deoxythymidine. Clin. Pharmacol. Ther. 1992, 51, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Balimane, P.V.; Sinko, P.J. Involvement of multiple transporters in the oral absorption of nucleoside analogues. Adv. Drug Deliv. Rev. 1999, 39, 183–209. [Google Scholar] [CrossRef]
- Grasela, D.M.; Stoltz, R.R.; Barry, M.; Bone, M.; Mangold, B.; O’Grady, P.; Raymond, R.; Haworth, S.J. Pharmacokinetics of single-dose oral stavudine in subjects with renal impairment and in subjects requiring hemodialysis. Antimicrob. Agents Chemother. 2000, 44, 2149–2153. [Google Scholar] [CrossRef][Green Version]
- Johnson, M.A.; Moore, K.H.; Yuen, G.J.; Bye, A.; Pakes, G.E. Clinical pharmacokinetics of lamivudine. Clin. Pharmacokinet. 1999, 36, 41–66. [Google Scholar] [CrossRef]
- Johnson, M.A.; Horak, J.; Breuel, P. The pharmacokinetics of lamivudine in patients with impaired hepatic function. Eur. J. Clin. Pharmacol. 1998, 54, 363–366. [Google Scholar] [CrossRef]
- McDowell, J.A.; Lou, Y.; Symonds, W.S.; Stein, D.S. Multiple-dose pharmacokinetics and pharmacodynamics of abacavir alone and in combination with zidovudine in human immunodeficiency virus-infected adults. Antimicrob. Agents Chemother. 2000, 44, 2061–2067. [Google Scholar] [CrossRef]
- Kearney, B.P.; Yale, K.; Shah, J.; Zhong, L.; Flaherty, J.F. Pharmacokinetics and dosing recommendations of tenofovir disoproxil fumarate in hepatic or renal impairment. Clin. Pharmacokinet. 2006, 45, 1115–1124. [Google Scholar] [CrossRef]
- Custodio, J.M.; Fordyce, M.; Garner, W.; Vimal, M.; Ling, K.H.J.; Kearney, B.P.; Ramanathan, S. Pharmacokinetics and safety of tenofovir alafenamide in HIV-uninfected subjects with severe renal impairment. Antimicrob. Agents Chemother. 2016, 60, 5135–5140. [Google Scholar] [CrossRef]
- Lamorde, M.; Byakika-Kibwika, P.; Tamale, W.S.; Kiweewa, F.; Ryan, M.; Amara, A.; Tjia, J.; Back, D.; Khoo, S.; Boffito, M.; et al. Effect of food on the steady-state pharmacokinetics of tenofovir and emtricitabine plus efavirenz in ugandan adults. AIDS Res. Treat. 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- To, K.W.; Liu, S.T.; Cheung, S.W.; Chan, D.P.C.; Chan, R.C.Y.; Lee, S.S. Pharmacokinetics of plasma efavirenz and CYP2B6 polymorphism in Southern Chinese. Ther. Drug Monit. 2009, 31, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Van Heeswijk, R.P.G.; Veldkamp, A.I.; Mulder, J.W.; Meenhorst, P.L.; Wit, F.W.N.M.; Lange, J.M.A.; Danner, S.A.; Foudraine, N.A.; Kwakkelstein, M.O.; Reiss, P.; et al. The steady-state pharmacokinetics of nevirapine during once daily and twice daily dosing in HIV-1-infected individuals. AIDS 2000, 14, F77–F82. [Google Scholar] [CrossRef] [PubMed]
- De Maat, M.M.R.; Huitema, A.D.R.; Mulder, J.W.; Meenhorst, P.L.; Van Gorp, E.C.M.; Beijnen, J.H. Population pharmacokinetics of nevirapine in an unselected cohort of HIV-1-infected individuals. Br. J. Clin. Pharmacol. 2002, 54, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.M.; Mulcahy, F.M.; Tjia, J.; Reynolds, H.E.; Gibbons, S.E.; Barry, M.G.; Back, D.J. Pharmacokinetic Interactions of Nevirapine and Methadone and Guidelines for Use of Nevirapine to Treat Injection Drug Users. Clin. Infect. Dis. 2001, 33, 1595–1597. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Battegay, M.; Arasteh, K.; Plettenberg, A.; Bogner, J.R.; Livrozet, J.M.; Witt, M.D.; Mossdorf, E.; Yong, C.L.; Zhang, W.; Macha, S.; et al. Bioavailability of Extended-Release Nevirapine 400 and 300 mg in HIV-1: A Multicenter, Open-Label Study. Clin. Ther. 2011, 33, 1308–1320. [Google Scholar] [CrossRef] [PubMed]
- Yong, C.L.; Gathe, J.C.; Knecht, G.; Orrell, C.; Mallolas, J.; Podzamczer, D.; Trottier, B.; Zhang, W.; Sabo, J.P.; Vinisko, R.; et al. Pharmacokinetic analysis of nevirapine extended release 400 mg once daily vs nevirapine immediate release 200 mg twice daily formulation in treatment-naïve patients with HIV-1 infection. HIV Clin. Trials 2017, 18, 189–195. [Google Scholar] [CrossRef]
- Schöller-Gyüre, M.; Kakuda, T.N.; De Smedt, G.; Woodfall, B.; Berckmans, C.; Peeters, M.; Hoetelmans, R.M.W. Effects of hepatic impairment on the steady-state pharmacokinetics of etravirine 200 mg BID: An open-label, multiple-dose, controlled Phase I study in adults. Clin. Ther. 2010, 32, 328–337. [Google Scholar] [CrossRef]
- John, J.; Liang, D. Oral liquid formulation of etravirine for enhanced bioavailability. J. Bioequivalence Bioavailab. 2014, 6, 46–52. [Google Scholar]
- Crauwels, H.M.; Van Heeswijk, R.P.G.; Buelens, A.; Stevens, M.; Boven, K.; Hoetelmans, R.M.W. Impact of food and different meal types on the pharmacokinetics of rilpivirine. J. Clin. Pharmacol. 2013, 53, 834–840. [Google Scholar] [CrossRef]
- Washington, C.B.; Flexner, C.; Sheiner, L.B.; Rosenkranz, S.L.; Segal, Y.; Aberg, J.A.; Blaschke, T.F. Effect of simultaneous versus staggered dosing on pharmacokinetic interactions of protease inhibitors. Clin. Pharmacol. Ther. 2003, 73, 406–416. [Google Scholar] [CrossRef]
- Hsu, A.; Isaacson, J.; Brun, S.; Bernstein, B.; Lam, W.; Bertz, R.; Foit, C.; Rynkiewicz, K.; Richards, B.; King, M.; et al. Pharmacokinetic-pharmacodynamic analysis of lopinavir-ritonavir in combination with efavirenz and two nucleoside reverse transcriptase inhibitors in extensively pretreated human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 2003, 47, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.C.; Deutsch, P.J.; Haddix, H.; Hesney, M.; Hoagland, V.; Ju, W.D.; Justice, S.J.; Osborne, B.; Sterrett, A.T.; Stone, J.A.; et al. Single-dose pharmacokinetics of indinavir and the effect of food. Antimicrob Agents Chemother 1998, 42, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Damle, B.; Hewlett, D.; Hsyu, P.H.; Becker, M.; Petersen, A. Pharmacokinetics of nelfinavir in subjects with hepatic impairment. J. Clin. Pharmacol. 2006, 46, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Musiime, V.; Fillekes, Q.; Kekitiinwa, A.; Kendall, L.; Keishanyu, R.; Namuddu, R.; Young, N.; Opilo, W.; Lallemant, M.; Walker, A.S.; et al. The pharmacokinetics and acceptability of lopinavir/ritonavir minitab sprinkles, tablets, and syrups in african HIV-infected children. J. Acquir. Immune Defic. Syndr. 2014, 66, 148–154. [Google Scholar] [CrossRef]
- Veronese, L.; Rautaureau, J.; Sadler, B.M.; Gillotin, C.; Petite, J.P.; Pillegand, B.; Delvaux, M.; Masliah, C.; Fosse, S.; Lou, Y.; et al. Single-dose pharmacokinetics of amprenavir, a human immunodeficiency virus type 1 protease inhibitor, in subjects with normal or impaired hepatic function. Antimicrob. Agents Chemother. 2000, 44, 821–826. [Google Scholar] [CrossRef]
- Sevinsky, H.; Tao, X.; Wang, R.; Ravindran, P.; Sims, K.; Xu, X.; Jariwala, N.; Bertz, R. A randomized trial in healthy subjects to assess the bioequivalence of an atazanavir/cobicistat fixed-dose combination tablet versus administration as separate agents. Antivir. Ther. 2015, 20, 493–500. [Google Scholar] [CrossRef]
- MacGregor, T.R.; Sabo, J.P.; Norris, S.H.; Johnson, P.; Galitz, L.; McCallister, S. Pharmacokinetic characterization of different dose combinations of coadministered tipranavir and ritonavir in healthy volunteers. HIV Clin. Trials 2004, 5, 371–382. [Google Scholar] [CrossRef]
- Sekar, V.; Kestens, D.; Spinosa-Guzman, S.; De Pauw, M.; De Paepe, E.; Vangeneugden, T.; Lefebvre, E.; Hoetelmans, R.M.W. The effect of different meal types on the pharmacokinetics of darunavir (TMC114)/ritonavir in HIV-negative healthy volunteers. J. Clin. Pharmacol. 2007, 47, 479–484. [Google Scholar] [CrossRef]
- Rittweger, M.; Arastéh, K. Clinical pharmacokinetics of darunavir. Clin. Pharmacokinet. 2007, 46, 739–756. [Google Scholar] [CrossRef]
- Kilby, J.M.; Lalezari, J.P.; Eron, J.J.; Carlson, M.; Cohen, C.; Arduino, R.C.; Goodgame, J.C.; Gallant, J.E.; Volberding, P.; Murphy, R.L.; et al. The safety, plasma pharmacokinetics, and antiviral activity of subcutaneous enfuvirtide (T-20), a peptide inhibitor of gp41-mediated virus fusion, in HIV-infected adults. AIDS Res. Hum. Retrovir. 2002, 18, 685–693. [Google Scholar] [CrossRef]
- Abel, S.; Russell, D.; Whitlock, L.A.; Ridgway, C.E.; Nedderman, A.N.R.; Walker, D.K. Assessment of the absorption, metabolism and absolute bioavailability of maraviroc in healthy male subjects. Br. J. Clin. Pharmacol. 2008, 65, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Song, I.; Borland, J.; Chen, S.; Patel, P.; Wajima, T.; Peppercorn, A.; Piscitelli, S.C. Effect of food on the pharmacokinetics of the integrase inhibitor dolutegravir. Antimicrob. Agents Chemother. 2012, 56, 1627–1629. [Google Scholar] [CrossRef] [PubMed]
- Moss, L.; Wagner, D.; Kanaoka, E.; Olson, K.; Yueh, Y.L.; Bowers, G.D. The comparative disposition and metabolism of dolutegravir, a potent HIV-1 integrase inhibitor, in mice, rats, and monkeys. Xenobiotica 2015, 45, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Mathias, A.A.; West, S.; Hui, J.; Kearney, B.P. Dose-response of ritonavir on hepatic CYP3A activity and elvitegravir oral exposure. Clin. Pharmacol. Ther. 2009, 85, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Assessment Report of Vitekta, EMA/701401/2013, European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/vitekta (accessed on 5 September 2019).
- Iwamoto, M.; Hanley, W.D.; Petry, A.S.; Friedman, E.J.; Kost, J.T.; Breidinger, S.A.; Lasseter, K.C.; Robson, R.; Lunde, N.M.; Wenning, L.A.; et al. Lack of a clinically important effect of moderate hepatic insufficiency and severe renal insufficiency on raltegravir pharmacokinetics. Antimicrob. Agents Chemother. 2009, 53, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Gallant, J.E.; Thompson, M.; DeJesus, E.; Voskuhl, G.W.; Wei, X.; Zhang, H.; White, K.; Cheng, A.; Quirk, E.; Martin, H. Antiviral activity, safety, and pharmacokinetics of bictegravir as 10-day monotherapy in HIV-1-infected adults. J. Acquir. Immune Defic. Syndr. 2017, 75, 61–66. [Google Scholar] [CrossRef]
- Gao, Y.; Kraft, J.C.; Yu, D.; Ho, R.J.Y. Recent developments of nanotherapeutics for targeted and long-acting, combination HIV chemotherapy. Eur. J. Pharm. Biopharm. 2019, 138, 75–91. [Google Scholar] [CrossRef]
- Devi, K.; Pai, R. Antiretrovirals: Need for an effective drug delivery. Indian J. Pharm. Sci. 2006, 68, 1–6. [Google Scholar] [CrossRef]
- Wuyts, B.; Brouwers, J.; Mols, R.; Tack, J.; Annaert, P.; Augustijns, P. Solubility profiling of HIV protease inhibitors in human intestinal fluids. J. Pharm. Sci. 2013, 102, 3800–3807. [Google Scholar] [CrossRef]
- Bastiaans, D.E.; Cressey, T.R.; Vromans, H.; Burger, D.M. The role of formulation on the pharmacokinetics of antiretroviral drugs. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1019–1037. [Google Scholar] [CrossRef]
- Amin, M.L. P-glycoprotein inhibition for optimal drug delivery. Drug Target. Insights 2013, 2013, 27–34. [Google Scholar] [CrossRef]
- Wilkinson, G.R. Cytochrome P4503A (CYP3A) metabolism: Prediction of in vivo activity in humans. J. Pharmacokinet. Biopharm. 1996, 24, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, C.M.; Buchanan, N.L.; Edgar, K.J.; Little, J.L.; Ramsey, M.G.; Ruble, K.M.; Wacher, V.J.; Wempe, M.F. Pharmacokinetics of saquinavir after intravenous and oral dosing of Saquinavir: Hydroxybutenyl-β-cyclodextrin formulations. Biomacromolecules 2008, 9, 305–313. [Google Scholar] [CrossRef]
- Swartz, M.A. The physiology of the lymphatic system. Adv. Drug Deliv. Rev. 2001, 50, 3–20. [Google Scholar] [CrossRef]
- Trevaskis, N.L.; Charman, W.N.; Porter, C.J.H. Lipid-based delivery systems and intestinal lymphatic drug transport: A mechanistic update. Adv. Drug Deliv. Rev. 2008, 60, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Yáñez, J.A.; Wang, S.W.J.; Knemeyer, I.W.; Wirth, M.A.; Alton, K.B. Intestinal lymphatic transport for drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 923–942. [Google Scholar] [CrossRef] [PubMed]
- Trezza, C.R.; Kashuba, A.D.M. Pharmacokinetics of antiretrovirals in genital secretions and anatomic sites of HIV transmission: Implications for HIV prevention. Clin. Pharmacokinet. 2014, 53, 611–624. [Google Scholar] [CrossRef]
- Palmer, S.; Maldarelli, F.; Wiegand, A.; Bernstein, B.; Hanna, G.J.; Brun, S.C.; Kempf, D.J.; Mellors, J.W.; Coffin, J.M.; King, M.S. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2008, 105, 3879–3884. [Google Scholar] [CrossRef]
- Kearney, M.F.; Wiegand, A.; Shao, W.; Coffin, J.M.; Mellors, J.W.; Lederman, M.; Gandhi, R.T.; Keele, B.F.; Li, J.Z. Origin of Rebound Plasma HIV Includes Cells with Identical Proviruses That Are Transcriptionally Active before Stopping of Antiretroviral Therapy. J. Virol. 2016, 90, 1369–1376. [Google Scholar] [CrossRef]
- Gunaseelan, S.; Gunaseelan, K.; Deshmukh, M.; Zhang, X.; Sinko, P.J. Surface modifications of nanocarriers for effective intracellular delivery of anti-HIV drugs. Adv. Drug Deliv. Rev. 2010, 62, 518–531. [Google Scholar] [CrossRef]
- Shao, J.; Kraft, J.C.; Li, B.; Yu, J.; Freeling, J.; Koehn, J.; Ho, R.J. Nanodrug formulations to enhance HIV drug exposure in lymphoid tissues and cells: Clinical significance and potential impact on treatment and eradication of HIV/AIDS. Futur. Med. 2016, 11, 545–564. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil®-The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Tejada-Berges, T.; Granai, C.O.; Gordinier, M.; Gajewski, W. Caelyx/Doxil for the treatment of metastatic ovarian and breast cancer. Expert Rev. Anticancer Ther. 2002, 2, 143–150. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.E.R.; Wigler, N.; Inbar, M.; Rosso, R.; Grischke, E.; Santoro, A.; Catane, R.; Kieback, D.G.; Tomczak, P.; Ackland, S.P.; et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYXTM/Doxil®) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann. Oncol. 2004, 15, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, V.; Etherington, A.; Greene, M.; Quijano, J.; Xie, J.; Hunter, F.; Dromi, S.; Li, K.C.P. Delivery of liposomal doxorubicin (Doxil) in a breast cancer tumor model: Investigation of potential enhancement by pulsed-high intensity focused ultrasound exposure. Acad. Radiol. 2006, 13, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Simpson, C.A.; Dahl, N.P.; Bresee, J.; Whitehead, D.C.; Lindsey, E.A.; Harris, T.L.; Smith, C.A.; Carter, C.J.; Feldheim, D.L.; et al. Gold nanoparticles to improve HIV drug delivery. Future Med. Chem. 2015, 7, 1097–1107. [Google Scholar] [CrossRef]
- Cao, S.; Woodrow, K.A. Nanotechnology approaches to eradicating HIV reservoirs. Eur. J. Pharm. Biopharm. 2019, 138, 48–63. [Google Scholar] [CrossRef]
- Lara, H.H.; Ixtepan-Turrent, L.; Garza Treviño, E.N.; Singh, D.K. Use of silver nanoparticles increased inhibition of cell-associated HIV-1 infection by neutralizing antibodies developed against HIV-1 envelope proteins. J. Nanobiotechnology 2011, 9, 38. [Google Scholar] [CrossRef]
- Chopra, S.; Venkatesan, N.; Betageri, G.V. Liposomes as nanocarriers for anti-HIV therapy. Drug Deliv. Transl. Res. 2013, 3, 471–478. [Google Scholar] [CrossRef]
- Kraft, J.C.; Freeling, J.P.; Wang, Z.; Ho, R.J.Y. Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J. Pharm. Sci. 2014, 103, 29–52. [Google Scholar] [CrossRef]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, P.; Hall, J.B.; McLeland, C.B.; Dobrovolskaia, M.A.; McNeil, S.E. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv. Drug Deliv. Rev. 2009, 61, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Gohla, S.; Keck, C.M. State of the art of nanocrystals-Special features, production, nanotoxicology aspects and intracellular delivery. Eur. J. Pharm. Biopharm. 2011, 78, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Owen, A.; Rannard, S. Strengths, weaknesses, opportunities and challenges for long acting injectable therapies: Insights for applications in HIV therapy. Adv. Drug Deliv. Rev. 2016, 103, 144–156. [Google Scholar] [CrossRef]
- Margolis, D.A.; Gonzalez-Garcia, J.; Stellbrink, H.J.; Eron, J.J.; Yazdanpanah, Y.; Podzamczer, D.; Lutz, T.; Angel, J.B.; Richmond, G.J.; Clotet, B.; et al. Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet 2017, 390, 1499–1510. [Google Scholar] [CrossRef]
- Whitfield, T.; Torkington, A.; van Halsema, C. Profile of cabotegravir and its potential in the treatment and prevention of HIV-1 infection: Evidence to date. HIV/AIDS Res. Palliat. Care 2016, 8, 157–164. [Google Scholar] [CrossRef]
- ViiV Healthcare submits New Drug Application to US FDA ViiV Healthcare. Available online: https://www.viivhealthcare.com/en-gb/media/press-releases/2019/april/viiv-healthcare-submits-new-drug-application-to-us-fda-for-the-first-monthly-injectable-two-drug-regimen-of-cabotegravir-and-rilpivirine-for-treatment-of-hiv/ (accessed on 22 August 2019).
- Yant, S.R.; Mulato, A.; Hansen, D.; Tse, W.C.; Niedziela-Majka, A.; Zhang, J.R.; Stepan, G.J.; Jin, D.; Wong, M.H.; Perreira, J.M.; et al. A highly potent long-acting small-molecule HIV-1 capsid inhibitor with efficacy in a humanized mouse model. Nat. Med. 2019, 25, 1377–1384. [Google Scholar] [CrossRef]
- Riber, C.F.; Smith, A.A.A.; Zelikin, A.N. Self-Immolative Linkers Literally Bridge Disulfide Chemistry and the Realm of Thiol-Free Drugs. Adv. Healthc. Mater. 2015, 4, 1–4. [Google Scholar] [CrossRef]
- Andersen, A.H.F.; Riber, C.F.; Zuwala, K.; Tolstrup, M.; Dagnæs-Hansen, F.; Denton, P.W.; Zelikin, A.N. Long-Acting, Potent Delivery of Combination Antiretroviral Therapy. ACS Macro Lett. 2018, 7, 587–591. [Google Scholar] [CrossRef]
- Danial, M.; Andersen, A.H.F.; Żuwała, K.; Cosson, S.; Riber, C.F.; Smith, A.A.A.; Tolstrup, M.; Moad, G.; Zelikin, A.N.; Postma, A. Triple Activity of Lamivudine Releasing Sulfonated Polymers against HIV-1. Mol. Pharm. 2016, 13, 2397–2410. [Google Scholar] [CrossRef]
- Hinton, T.M.; Zuwala, K.; Deffrasnes, C.; Todd, S.; Shi, S.; Marsh, G.A.; Dearnley, M.; Wohl, B.M.; Tolstrup, M.; Zelikin, A.N. Polyanionic Macromolecular Prodrugs of Ribavirin: Antiviral Agents with a Broad Spectrum of Activity. Adv. Healthc. Mater. 2016, 5, 534–540. [Google Scholar] [CrossRef]
- Kock, A.; Zuwala, K.; Smith, A.A.A.; Ruiz-Sanchis, P.; Wohl, B.M.; Tolstrup, M.; Zelikin, A.N. Disulfide reshuffling triggers the release of a thiol-free anti-HIV agent to make up fast-acting, potent macromolecular prodrugs. Chem. Commun. 2014, 50, 14498–14500. [Google Scholar] [CrossRef] [PubMed]
- Zuwala, K.; Smith, A.A.A.; Tolstrup, M.; Zelikin, A.N. HIV anti-latency treatment mediated by macromolecular prodrugs of histone deacetylase inhibitor, panobinostat. Chem. Sci. 2016, 7, 2353–2358. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.A.A.; Zuwala, K.; Pilgram, O.; Johansen, K.S.; Tolstrup, M.; Dagnæs-Hansen, F.; Zelikin, A.N. Albumin-Polymer-Drug Conjugates: Long Circulating, High Payload Drug Delivery Vehicles. ACS Macro Lett. 2016, 5, 1089–1094. [Google Scholar] [CrossRef]
- O’Hagan, D.; Singh, M.; Ugozzoli, M.; Wild, C.; Barnett, S.; Chen, M.; Schaefer, M.; Doe, B.; Otten, G.R.; Ulmer, J.B. Induction of Potent Immune Responses by Cationic Microparticles with Adsorbed Human Immunodeficiency Virus DNA Vaccines. J. Virol. 2001, 75, 9037–9043. [Google Scholar] [CrossRef]
- Neeraj, A.; Chandrasekar, M.J.N.; Sara, U.V.S.; Rohini, A. Poly (HEMA-Zidovudine) conjugate: A macromolecular pro-drug for improvement in the biopharmaceutical properties of the drug. Drug Deliv. 2011, 18, 272–280. [Google Scholar] [CrossRef]
- Schandock, F.; Riber, C.F.; Röcker, A.; Müller, J.A.; Harms, M.; Gajda, P.; Zuwala, K.; Andersen, A.H.F.; Løvschall, K.B.; Tolstrup, M.; et al. Macromolecular Antiviral Agents against Zika, Ebola, SARS, and Other Pathogenic Viruses. Adv. Healthc. Mater. 2017, 6, 1700748. [Google Scholar] [CrossRef]
- Li, W.; Wang, Q.; Li, Y.; Yu, F.; Liu, Q.; Qin, B.; Xie, L.; Lu, L.; Jiang, S. A Nanoparticle-Encapsulated Non-Nucleoside Reverse-Transcriptase Inhibitor with Enhanced Anti-HIV-1 Activity and Prolonged Circulation Time in Plasma. Curr. Pharm. Des. 2014, 21, 925–935. [Google Scholar] [CrossRef]
- Frich, C.K.; Krüger, F.; Walther, R.; Domar, C.; Andersen, A.H.F.; Tvilum, A.; Dagnæs-Hansen, F.; Denton, P.W.; Tolstrup, M.; Paludan, S.R.; et al. Non-covalent hitchhiking on endogenous carriers as a protraction mechanism for antiviral macromolecular prodrugs. J. Control. Release 2019, 294, 298–310. [Google Scholar] [CrossRef]
- Chaudhury, C.; Mehnaz, S.; Robinson, J.M.; Hayton, W.L.; Pearl, D.K.; Roopenian, D.C.; Anderson, C.L. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J. Exp. Med. 2003, 197, 315–322. [Google Scholar] [CrossRef]
- Lau, J.; Bloch, P.; Schäffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B.; et al. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J. Med. Chem. 2015, 58, 7370–7380. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Moynihan, K.D.; Zheng, Y.; Szeto, G.L.; Li, A.V.; Huang, B.; Van Egeren, D.S.; Park, C.; Irvine, D.J. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 2014, 507, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yu, F.; Wang, Q.; Qi, Q.; Su, S.; Xie, L.; Lu, L.; Jiang, S. Co-delivery of HIV-1 entry inhibitor and nonnucleoside reverse transcriptase inhibitor shuttled by nanoparticles: Cocktail therapeutic strategy for antiviral therapy. AIDS 2016, 30, 827–837. [Google Scholar] [CrossRef]
- Mandal, S.; Belshan, M.; Holec, A.; Zhou, Y.; Destache, C.J. An Enhanced Emtricitabine-Loaded Long-Acting Nanoformulation for Prevention or Treatment of HIV Infection. Antimicrob. Agents Chemother. 2017, 61, e01475–e01516. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Kang, G.; Prathipati, P.K.; Zhou, Y.; Fan, W.; Li, Q.; Destache, C.J. Nanoencapsulation introduces long-acting phenomenon to tenofovir alafenamide and emtricitabine drug combination: A comparative pre-exposure prophylaxis efficacy study against HIV-1 vaginal transmission. J. Control. Release 2019, 294, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Prathipati, P.K.; Kang, G.; Zhou, Y.; Yuan, Z.; Fan, W.; Li, Q.; Destache, C.J. Tenofovir alafenamide and elvitegravir loaded nanoparticles for long-acting prevention of HIV-1 vaginal transmission. AIDS 2017, 31, 469–476. [Google Scholar] [CrossRef]
- Destache, C.J.; Belgum, T.; Goede, M.; Shibata, A.; Belshan, M.A. Antiretroviral release from poly(DL-lactide-co-glycolide) nanoparticles in mice. J. Antimicrob. Chemother. 2010, 65, 2183–2187. [Google Scholar] [CrossRef]
- Guo, D.; Zhang, G.; Wysocki, T.A.; Wysocki, B.J.; Gelbard, H.A.; Liu, X.-M.; McMillan, J.M.; Gendelman, H.E. Endosomal Trafficking of Nanoformulated Antiretroviral Therapy Facilitates Drug Particle Carriage and HIV Clearance. J. Virol. 2014, 88, 9504–9513. [Google Scholar] [CrossRef]
- Williams, J.; Sayles, H.R.; Meza, J.L.; Sayre, P.; Sandkovsky, U.; Gendelman, H.E.; Flexner, C.; Swindells, S. Long-acting parenteral nanoformulated antiretroviral therapy: Interest and attitudes of HIV-infected patients. Nanomedicine 2013, 8, 1807–1813. [Google Scholar] [CrossRef]
- Edagwa, B.; McMillan, J.E.; Sillman, B.; Gendelman, H.E. Long-acting slow effective release antiretroviral therapy. Expert Opin. Drug Deliv. 2017, 14, 1281–1291. [Google Scholar] [CrossRef]
- Sillman, B.; Bade, A.N.; Dash, P.K.; Bhargavan, B.; Kocher, T.; Mathews, S.; Su, H.; Kanmogne, G.D.; Poluektova, L.Y.; Gorantla, S.; et al. Creation of a long-acting nanoformulated dolutegravir. Nat. Commun. 2018, 9, 443. [Google Scholar] [CrossRef] [PubMed]
- Edagwa, B.; Zhou, T.; McMillan, J.; Liu, X.-M.; Gendelman, H. Development of HIV Reservoir Targeted Long Acting Nanoformulated Antiretroviral Therapies. Curr. Med. Chem. 2014, 21, 4186–4198. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.K.; Kaminski, R.; Bella, R.; Su, H.; Mathews, S.; Ahooyi, T.M.; Chen, C.; Mancuso, P.; Sariyer, R.; Ferrante, P.; et al. Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat. Commun. 2019, 10, 2753. [Google Scholar] [CrossRef] [PubMed]
- Golombek, S.K.; May, J.N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar] [CrossRef]
- Richter, W.F.; Bhansali, S.G.; Morris, M.E.; Svensson, C.; Balthasar, J.; Theil, F.-P.; Richter, W.F.; Bhansali, S.G.; Morris, M.E. Mechanistic determinants of biotherapeutics absorption following SC administration. AAPS J. 2012, 14, 559–570. [Google Scholar] [CrossRef]
- Kinman, L.; Bui, T.; Larsen, K.; Tsai, C.-C.; Anderson, D.; Morton, W.R.; Hu, S.; Ho, R.J.Y. Optimization of Lipid Y Indinavir Complexes for Localization in Lymphoid Tissues of HIV-Infected Macaques. J. Acquir. Immune Defic. Syndr. 2006, 42, 155–161. [Google Scholar] [CrossRef]
- Kinman, L.; Brodie, S.J.; Tsai, C.C.; Bui, T.; Larsen, K.; Schmidt, A.; Anderson, D.; Morton, W.R.; Hu, S.L.; Ho, R.J.Y. Lipid-Drug Association Enhanced HIV-1 Protease Inhibitor Indinavir Localization in Lymphoid Tissues and Viral Load Reduction: A Proof of Concept Study in HIV-2287-Infected Macaques. J. Acquir. Immune Defic. Syndr. 2003, 34, 387–397. [Google Scholar] [CrossRef]
- Freeling, J.P.; Ho, R.J.Y. Anti-HIV drug particles may overcome lymphatic drug insufficiency and associated HIV persistence. Proc. Natl. Acad. Sci. USA 2014, 111, E2512–E2513. [Google Scholar] [CrossRef]
- Freeling, J.P.; Koehn, J.; Shu, C.; Sun, J.; Ho, R.J.Y. Anti-HIV Drug-Combination Nanoparticles Enhance Plasma Drug Exposure Duration as Well as Triple-Drug Combination Levels in Cells Within Lymph Nodes and Blood in Primates. AIDS Res. Hum. Retrovir. 2015, 31, 107–114. [Google Scholar] [CrossRef]
- Kraft, J.C.; Mcconnachie, L.A.; Koehn, J.; Kinman, L.; Collins, C.; Shen, D.D.; Collier, A.C.; Ho, R.J.Y. Long-acting combination anti-HIV drug suspension enhances and sustains higher drug levels in lymph node cells than in blood cells and plasma. AIDS 2017, 31, 765–770. [Google Scholar] [CrossRef]
- Kraft, J.C.; Treuting, P.M.; Ho, R.J.Y. Indocyanine green nanoparticles undergo selective lymphatic uptake, distribution and retention and enable detailed mapping of lymph vessels, nodes and abnormalities. J. Drug Target. 2018, 26, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Kraft, J.C.; McConnachie, L.A.; Koehn, J.; Kinman, L.; Sun, J.; Collier, A.C.; Collins, C.; Shen, D.D.; Ho, R.J.Y. Mechanism-based pharmacokinetic (MBPK) models describe the complex plasma kinetics of three antiretrovirals delivered by a long-acting anti-HIV drug combination nanoparticle formulation. J. Control. Release 2018, 275, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Glass, J.J.; Yuen, D.; Rae, J.; Johnston, A.P.R.; Parton, R.G.; Kent, S.J.; De Rose, R. Human immune cell targeting of protein nanoparticles-caveospheres. Nanoscale 2016, 8, 8255–8265. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Chen, Y.; Kaien, G.; Dash, A.; Sayre, C.L.; Davies, N.M.; Ho, E.A. Novel intravaginal nanomedicine for the targeted delivery of saquinavir to CD + immune cells. Int. J. Nanomed. 2013, 8, 2847–2858. [Google Scholar]
- Ramana, L.N.; Sharma, S.; Sethuraman, S.; Ranga, U.; Krishnan, U.M. Stealth anti-CD4 conjugated immunoliposomes with dual antiretroviral drugs-Modern Trojan horses to combat HIV. Eur. J. Pharm. Biopharm. 2015, 89, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Kovochich, M.; Marsden, M.D.; Zack, J.A. Activation of Latent HIV Using Drug-Loaded Nanoparticles. PLoS ONE 2011, 6, e18270. [Google Scholar] [CrossRef]
- Endsley, A.N.; Ho, R.J.Y. Enhanced anti-HIV efficacy of indinavir after inclusion in CD4-targeted lipid nanoparticles. J. Acquir. Immune Defic. Syndr. 2012, 61, 417–424. [Google Scholar] [CrossRef]
- Endsley, A.N.; Ho, R.J.Y. Design and characterization of novel peptide-coated lipid nanoparticles for targeting anti-HIV drug to CD4 expressing cells. AAPS J. 2012, 14, 225–235. [Google Scholar] [CrossRef]
- Pelchen-Matthews, A.; Clapham, P.; Marsh, M. Role of CD4 endocytosis in human immunodeficiency virus infection. J. Virol. 1995, 69, 8164–8168. [Google Scholar] [CrossRef]
- Pelchen-Matthews, A.; Armes, J.E.; Griffiths, G.; Marsh, M. Differential endocytosis of CD4 in lymphocytic and nonlymphocytic cells. J. Exp. Med. 1991, 173, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Pollock, S.; Dwek, R.A.; Burton, D.R.; Zitzmann, N. N-Butyldeoxynojirimycin is a broadly effective anti-HIV therapy significantly enhanced by targeted liposome delivery. AIDS 2008, 22, 1961–1969. [Google Scholar] [CrossRef] [PubMed]
- Descours, B.; Petitjean, G.; López-Zaragoza, J.-L.; Bruel, T.; Raffel, R.; Psomas, C.; Reynes, J.; Lacabaratz, C.; Levy, Y.; Schwartz, O.; et al. CD32a is a marker of a CD4 T-cell HIV reservoir harbouring replication-competent proviruses. Nature 2017, 543, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Kootstra, N.A.; Hooibrink, B.; van Montfort, T.; Maurer, I.; Groen, K.; Jurriaans, S.; Bakker, M.; van Lint, C.; Berkhout, B.; et al. CD32+CD4+ T Cells Are Highly Enriched for HIV DNA and Can Support Transcriptional Latency. Cell Rep. 2020, 30, 2284–2296. [Google Scholar] [CrossRef]
- Bertagnolli, L.N.; White, J.A.; Simonetti, F.R.; Beg, S.A.; Lai, J.; Tomescu, C.; Murray, A.J.; Antar, A.A.R.; Zhang, H.; Margolick, J.B.; et al. The role of CD32 during HIV-1 infection. Nature 2018, 561, E17–E19. [Google Scholar] [CrossRef]
- Osuna, C.E.; Lim, S.Y.; Kublin, J.L.; Apps, R.; Chen, E.; Mota, T.M.; Huang, S.H.; Ren, Y.; Bachtel, N.D.; Tsibris, A.M.; et al. Evidence that CD32a does not mark the HIV-1 latent reservoir. Nature 2018, 561, E20–E28. [Google Scholar] [CrossRef]
- Pérez, L.; Anderson, J.; Chipman, J.; Thorkelson, A.; Chun, T.W.; Moir, S.; Haase, A.T.; Douek, D.C.; Schacker, T.W.; Boritz, E.A. Conflicting evidence for HIV enrichment in CD32 + CD4 T cells. Nature 2018, 561, E9–E16. [Google Scholar] [CrossRef]
- Abdel-Mohsen, M.; Kuri-Cervantes, L.; Grau-Exposito, J.; Spivak, A.M.; Nell, R.A.; Tomescu, C.; Vadrevu, S.K.; Giron, L.B.; Serra-Peinado, C.; Genescà, M.; et al. CD32 is expressed on cells with transcriptionally active HIV but does not enrich for HIV DNA in resting T cells. Sci. Transl. Med. 2018, 10, eaar6759. [Google Scholar] [CrossRef]
- Martin, G.E.; Pace, M.; Thornhill, J.P.; Phetsouphanh, C.; Meyerowitz, J.; Gossez, M.; Brown, H.; Olejniczak, N.; Lwanga, J.; Ramjee, G.; et al. CD32-expressing CD4 T cells are phenotypically diverse and can contain proviral HIV DNA. Front. Immunol. 2018, 9, 928. [Google Scholar] [CrossRef]
- Prator, C.A.; Thanh, C.; Kumar, S.; Pan, T.; Peluso, M.J.; Bosch, R.; Jones, N.; Milush, J.M.; Bakkour, S.; Stone, M.; et al. Circulating CD30+ CD4+ T Cells Increase Prior to HIV Rebound Following Analytical Antiretroviral Treatment Interruption. J. Infect. Dis. 2019, 221, 1146–1155. [Google Scholar] [CrossRef]
- Wang, C.C.; Thanh, C.; Gibson, E.A.; Ball-Burack, M.; Hogan, L.E.; Descours, B.; Jones, N.; Carvidi, A.B.; Munter, S.; Bakkour, S.; et al. Transient loss of detectable HIV-1 RNA following brentuximab vedotin anti-CD30 therapy for Hodgkin lymphoma. Blood Adv. 2018, 2, 3479–3482. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.A.; Lewin, S.R. Shocking HIV out of hiding: Where are we with clinical trials of latency reversing agents? Curr. Opin. HIV AIDS 2016, 11, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Jayant, R.D.; Atluri, V.S.R.; Agudelo, M.; Sagar, V.; Kaushik, A.; Nair, M. Sustained-release nanoART formulation for the treatment of neuroAIDS. Int. J. Nanomed. 2015, 10, 1077–1093. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Liang, Y.; Liu, X.; Zhou, S.; Liu, L.; Zhang, F.; Xie, C.; Cai, S.; Wei, J.; Zhu, Y.; et al. PLGA-PEG Nanoparticles Coated with Anti-CD45RO and Loaded with HDAC Plus Protease Inhibitors Activate Latent HIV and Inhibit Viral Spread. Nanoscale Res. Lett. 2015, 10, 413. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering crispr: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug Class | Name (Acronym) | Plasma T½ in Humans (h) | Absolute Bioavailability (Fabs) |
|---|---|---|---|
| Nucleoside/nucleotide reverse transcriptase inhibitor (NRTIs) | Zidovudine (ZDV/AZT) [13] | 1.2 | 60–70% |
| Didanosine (DDI) [14] | 1.5 | 25–43% | |
| Stavudine (d4T) [15] | 1.6 | 82–99% | |
| Lamivudine (3TC) [16,17] | 5.4 | 86–88% | |
| Abacavir (ABC) [18] | 1.3 | 83% | |
| Tenofovir disoproxil fumarate (TDF) [19] | 18.3 | 25% in fasting, increased with food | |
| Tenofovir alafenamide (TAF) [20] | 51.3 | n/a | |
| Emtricitabine (FTC) [21] | 4.8 | ~100% | |
| Non-nucleoside reverse transcriptase inhibitors (NNRTIs) | Efavirenz (EFV) [22] | 37.7 | ~100% |
| Nevirapine (NVP) [23,24,25,26] | 21.5 | 90–93% | |
| Extended-release NVP [27] | 45 | n/a | |
| Etravirine (ETR) [28,29] | 30–50 | n/a | |
| Rilpivirine (RPV) [30] | 48 | n/a | |
| Protease inhibitors (PIs) | Saquinavir (SQV) [31] | 3.6 | 4–12% |
| Ritonavir (RTV) [32] | 3.5 | 60% | |
| Indinavir (IDV) [33] | 1.8 | ~100% | |
| Nelfinavir (NFV) [31,34] | 4.3 | ~100% | |
| Lopinavir (LPV) [35] | 5–6 | n/a | |
| Lopinavir (LPV) oral pellets | 5–6 | n/a | |
| Fosamprenavir (FPV) [36] | 4.8 | n/a | |
| Atazanavir (ATV) [37] | 7.5 | Low | |
| Tipranavir (TPV) [38] | 2.6 | n/a | |
| Darunavir (DRV) [39,40] | 14.6 | 37% (w/o ritonavir), 82% (with ritonavir) | |
| Fusion inhibitors | Enfurvirtide (T-20) [41] | 2 | n/a |
| Entry inhibitors | Maraviroc (MVC) [42] | 23 | 23.1–33% |
| Integrase inhibitors (INIs) | Dolutegravir (DTG) [43,44] | 13.5 | 87% (in monkeys) |
| Elvitegravir (EVG) [45,46] | 9.9 | <25% | |
| Raltegravir (RAL) [47] | 9.3 | n/a | |
| Bictegravir (BIC) [48] | 17.3 | n/a |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halling Folkmar Andersen, A.; Tolstrup, M. The Potential of Long-Acting, Tissue-Targeted Synthetic Nanotherapy for Delivery of Antiviral Therapy Against HIV Infection. Viruses 2020, 12, 412. https://doi.org/10.3390/v12040412
Halling Folkmar Andersen A, Tolstrup M. The Potential of Long-Acting, Tissue-Targeted Synthetic Nanotherapy for Delivery of Antiviral Therapy Against HIV Infection. Viruses. 2020; 12(4):412. https://doi.org/10.3390/v12040412
Chicago/Turabian StyleHalling Folkmar Andersen, Anna, and Martin Tolstrup. 2020. "The Potential of Long-Acting, Tissue-Targeted Synthetic Nanotherapy for Delivery of Antiviral Therapy Against HIV Infection" Viruses 12, no. 4: 412. https://doi.org/10.3390/v12040412
APA StyleHalling Folkmar Andersen, A., & Tolstrup, M. (2020). The Potential of Long-Acting, Tissue-Targeted Synthetic Nanotherapy for Delivery of Antiviral Therapy Against HIV Infection. Viruses, 12(4), 412. https://doi.org/10.3390/v12040412