Genetic Strain Diversity of Multi-Host RNA Viruses that Infect a Wide Range of Pollinators and Associates is Shaped by Geographic Origins


Abstract
1. Introduction
2. Materials and Methods
2.1. Specimen Collections
2.2. RNA Extraction and Reverse Transcription PCR for Virus Detection
2.3. Negative Strand Detection
2.4. Statistical Analyses
2.5. Phylogenetic Analyses
2.6. Phylogeny–Trait Correlation
3. Results
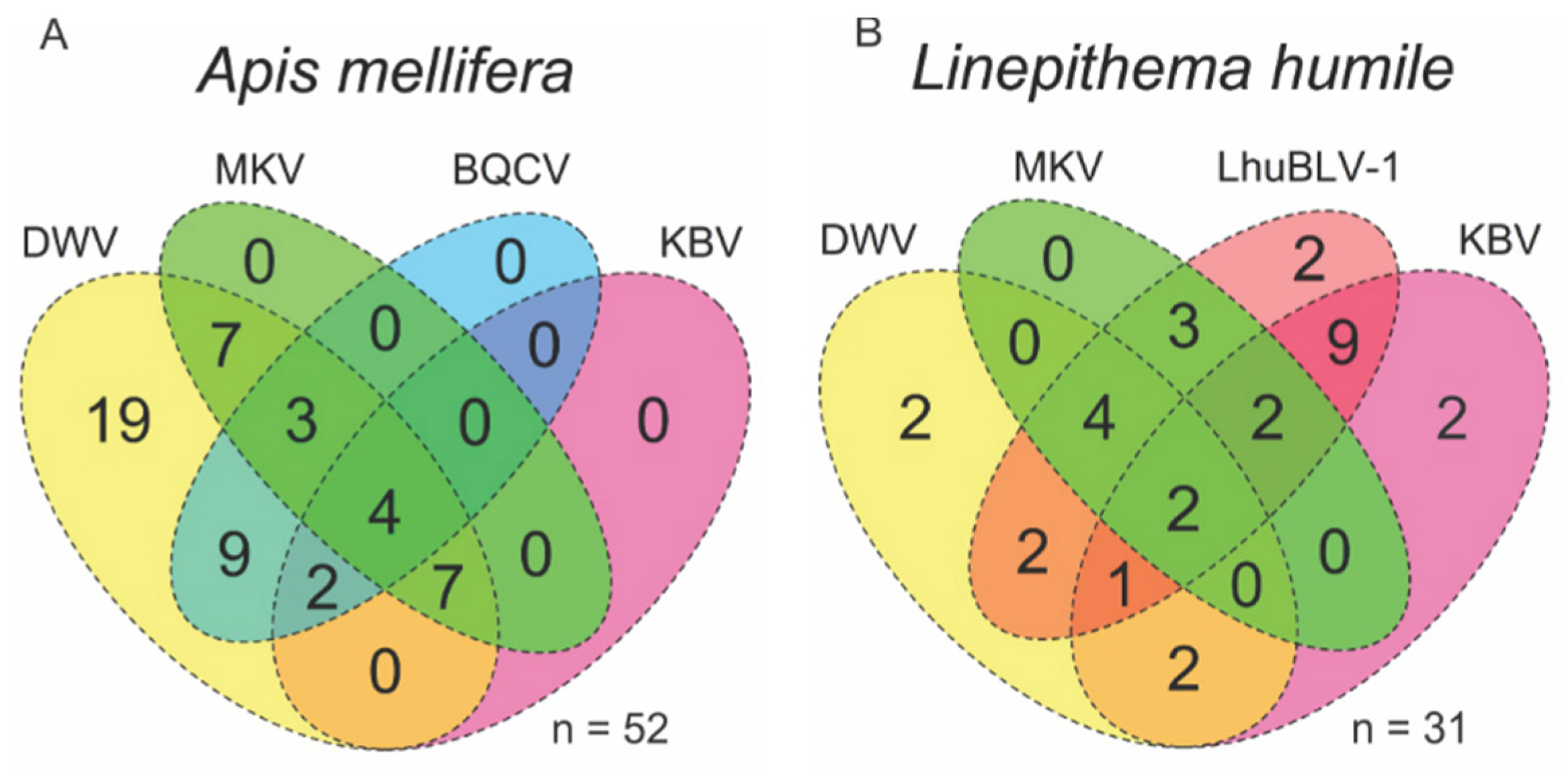
3.1. Viral Prevalence in Pollinators and Associated Arthropods
3.2. Viral Coinfections in Honey Bees and Argentine Ants
3.3. Viral Replication within Host Species
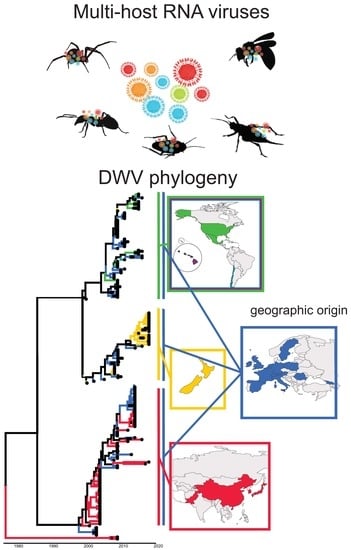
3.4. Viral Strain Diversity and Phylogenetic Analysis
3.5. Trait–Tip Associations
4. Discussion
4.1. Host Range of RNA Viruses
4.2. Pathogen Reservoir and Viral Spill Over into Wild Populations
4.3. Coinfections and Interactions among Viruses
4.4. Multi-Host Viruses in Emerging Disease
4.5. Global Distribution and Evolution of Bee Viruses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Potts, S.G.; Biesmeijer, J.C.; Kremen, C.; Neumann, P.; Schweiger, O.; Kunin, W.E. Global pollinator declines: Trends, impacts and drivers. Trends Ecol. Evol. 2010, 25, 345–353. [Google Scholar] [CrossRef]
- Hallmann, C.A.; Sorg, M.; Jongejans, E.; Siepel, H.; Hofland, N.; Schwan, H.; Stenmans, W.; Muller, A.; Sumser, H.; Horren, T.; et al. More than 75 percent decline over 27 years in total flying insect biomass in protected areas. PLoS ONE 2017, 12, e0185809. [Google Scholar] [CrossRef]
- Sánchez-Bayo, F.; Wyckhuys, K.A.G. Worldwide decline of the entomofauna: A review of its drivers. Biol. Conserv. 2019, 232, 8–27. [Google Scholar] [CrossRef]
- Goulson, D.; Nicholls, E.; Botías, C.; Rotheray, E.L. Bee declines driven by combined stress from parasites, pesticides, and lack of flowers. Science 2015, 347, 1255957. [Google Scholar] [CrossRef]
- Gallai, N.; Salles, J.M.; Settele, J.; Vaissiere, B.E. Economic valuation of the vulnerability of world agriculture confronted with pollinator decline. Ecol. Econ. 2009, 68, 810–821. [Google Scholar] [CrossRef]
- Manley, R.; Boots, M.; Wilfert, L. Emerging viral disease risk to pollinating insects: Ecological, evolutionary and anthropogenic factors. J Appl Ecol 2015, 52, 331–340. [Google Scholar] [CrossRef]
- Garibaldi, L.A.; Steffan-Dewenter, I.; Winfree, R.; Aizen, M.A.; Bommarco, R.; Cunningham, S.A.; Kremen, C.; Carvalheiro, L.G.; Harder, L.D.; Afik, O.; et al. Wild pollinators enhance fruit set of crops regardless of honey bee abundance. Science 2013, 339, 1608–1611. [Google Scholar] [CrossRef]
- Evans, J.D.; Schwarz, R.S. Bees brought to their knees: Microbes affecting honey bee health. Trends Microbiol. 2011, 19, 614–620. [Google Scholar] [CrossRef]
- McMenamin, A.J.; Genersch, E. Honey bee colony losses and associated viruses. Curr. Opin. Insect Sci. 2015, 8, 121–129. [Google Scholar] [CrossRef]
- Singh, R.; Levitt, A.L.; Rajotte, E.G.; Holmes, E.C.; Ostiguy, N.; Vanengelsdorp, D.; Lipkin, W.I.; Depamphilis, C.W.; Toth, A.L.; Cox-Foster, D.L. Rna viruses in hymenopteran pollinators: Evidence of inter-taxa virus transmission via pollen and potential impact on non-apis hymenopteran species. PLoS ONE 2010, 5, e14357. [Google Scholar] [CrossRef]
- Evison, S.E.; Roberts, K.E.; Laurenson, L.; Pietravalle, S.; Hui, J.; Biesmeijer, J.C.; Smith, J.E.; Budge, G.; Hughes, W.O. Pervasiveness of parasites in pollinators. PLoS ONE 2012, 7, e30641. [Google Scholar] [CrossRef]
- Murray, E.A.; Burand, J.; Trikoz, N.; Schnabel, J.; Grab, H.; Danforth, B.N. Viral transmission in honey bees and native bees, supported by a global black queen cell virus phylogeny. Environ. Microbiol. 2019, 21, 972–983. [Google Scholar] [CrossRef]
- Levitt, A.L.; Singh, R.; Cox-Foster, D.L.; Rajotte, E.; Hoover, K.; Ostiguy, N.; Holmes, E.C. Cross-species transmission of honey bee viruses in associated arthropods. Virus Res. 2013, 176, 232–240. [Google Scholar] [CrossRef]
- Schläppi, D.; Lattrell, P.; Yañez, O.; Chejanovsky, N.; Neumann, P. Foodborne transmission of deformed wing virus to ants (myrmica rubra). Insects 2019, 10, 394. [Google Scholar] [CrossRef]
- Sebastien, A.; Lester, P.J.; Hall, R.J.; Wang, J.; Moore, N.E.; Gruber, M.A. Invasive ants carry novel viruses in their new range and form reservoirs for a honeybee pathogen. Biol. Lett. 2015, 11, 20150610. [Google Scholar] [CrossRef]
- Brettell, L.E.; Schroeder, D.C.; Martin, S.J. Rnaseq analysis reveals virus diversity within hawaiian apiary insect communities. Viruses 2019, 11, 397. [Google Scholar] [CrossRef]
- Bailes, E.J.; Deutsch, K.R.; Bagi, J.; Rondissone, L.; Brown, M.J.F.; Lewis, O.T. First detection of bee viruses in hoverfly (syrphid) pollinators. Biol. Lett. 2018, 14. [Google Scholar] [CrossRef]
- Santamaria, J.; Villalobos, E.M.; Brettell, L.E.; Nikaido, S.; Graham, J.R.; Martin, S. Evidence of varroa-mediated deformed wing virus spillover in hawaii. J. Invertebr. Pathol. 2018, 151, 126–130. [Google Scholar] [CrossRef]
- Alger, S.A.; Burnham, P.A.; Boncristiani, H.F.; Brody, A.K. Rna virus spillover from managed honeybees (apis mellifera) to wild bumblebees (bombus spp.). PLoS ONE 2019, 14, e0217822. [Google Scholar] [CrossRef]
- Fürst, M.A.; McMahon, D.P.; Osborne, J.L.; Paxton, R.J.; Brown, M.J. Disease associations between honeybees and bumblebees as a threat to wild pollinators. Nature 2014, 506, 364–366. [Google Scholar] [CrossRef]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of rna viruses in arthropods reveals the ancestry of negative-sense rna viruses. eLife 2015, 4. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate rna virosphere. Nature 2016, 540, 539. [Google Scholar] [CrossRef]
- Woolhouse, M.E.J.; Haydon, D.T.; Antia, R. Emerging pathogens: The epidemiology and evolution of species jumps. Trends Ecol. Evol. 2005, 20, 238–244. [Google Scholar] [CrossRef]
- Johnson, P.T.J.; de Roode, J.C.; Fenton, A. Why infectious disease research needs community ecology. Science 2015, 349, 1259504. [Google Scholar] [CrossRef]
- Tehel, A.; Brown, M.J.; Paxton, R.J. Impact of managed honey bee viruses on wild bees. Curr. Opin. Virol. 2016, 19, 16–22. [Google Scholar] [CrossRef]
- Holmes, E.C. The evolutionary genetics of emerging viruses. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 353–372. [Google Scholar] [CrossRef]
- Figueroa, L.L.; Blinder, M.; Grincavitch, C.; Jelinek, A.; Mann, E.K.; Merva, L.A.; Metz, L.E.; Zhao, A.Y.; Irwin, R.E.; McArt, S.H.; et al. Bee pathogen transmission dynamics: Deposition, persistence and acquisition on flowers. Proc. Biol. Sci. 2019, 286, 20190603. [Google Scholar] [CrossRef]
- Faillace, C.A.; Lorusso, N.S.; Duffy, S. Overlooking the smallest matter: Viruses impact biological invasions. Ecol. Lett. 2017, 20, 524–538. [Google Scholar] [CrossRef]
- Martin, S.J.; Highfield, A.C.; Brettell, L.; Villalobos, E.M.; Budge, G.E.; Powell, M.; Nikaido, S.; Schroeder, D.C. Global honey bee viral landscape altered by a parasitic mite. Science 2012, 336, 1304–1306. [Google Scholar] [CrossRef]
- Loope, K.J.; Baty, J.W.; Lester, P.J.; Wilson Rankin, E.E. Pathogen shifts in a honeybee predator following the arrival of the varroa mite. Proc. Biol. Sci. 2019, 286, 20182499. [Google Scholar] [CrossRef]
- Cremer, S. Pathogens and disease defense of invasive ants. Curr. Opin. Insect Sci. 2019, 33, 63–68. [Google Scholar] [CrossRef]
- Gruber, M.A.M.; Cooling, M.; Baty, J.W.; Buckley, K.; Friedlander, A.; Quinn, O.; Russell, J.; Sebastien, A.; Lester, P.J. Single-stranded rna viruses infecting the invasive argentine ant, linepithema humile. Sci. Rep. 2017, 7, 3304. [Google Scholar] [CrossRef]
- Buys, B. Relationships between argentine ants and honeybees in south africa. Relatsh Argent. Ants Honeybees S. Afr. 1990, 519–524. [Google Scholar]
- Galbraith, D.A.; Fuller, Z.L.; Ray, A.M.; Brockmann, A.; Frazier, M.; Gikungu, M.W.; Martinez, J.F.I.; Kapheim, K.M.; Kerby, J.T.; Kocher, S.D.; et al. Investigating the viral ecology of global bee communities with high-throughput metagenomics. Sci. Rep. 2018, 8, 8879. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Flenniken, M.L. Bee viruses: Ecology, pathogenicity, and impacts. Annu. Rev. Entomol. 2019, 64, 205–226. [Google Scholar] [CrossRef]
- Mordecai, G.J.; Brettell, L.E.; Pachori, P.; Villalobos, E.M.; Martin, S.J.; Jones, I.M.; Schroeder, D.C. Moku virus; a new iflavirus found in wasps, honey bees and varroa. Sci. Rep. 2016, 6, 34983. [Google Scholar] [CrossRef]
- Viljakainen, L.; Holmberg, I.; Abril, S.; Jurvansuu, J. Viruses of invasive argentine ants from the european main supercolony: Characterization, interactions and evolution. J. Gen. Virol. 2018, 99, 1129–1140. [Google Scholar] [CrossRef]
- Evans, J.D.; Schwarz, R.S.; Chen, Y.P.; Budge, G.; Cornman, R.S.; De la Rua, P.; de Miranda, J.R.; Foret, S.; Foster, L.; Gauthier, L.; et al. Standard methods for molecular research in apis mellifera. J. Apic. Res. 2013, 52, 1–54. [Google Scholar] [CrossRef]
- Yue, C.; Genersch, E. Rt-pcr analysis of deformed wing virus in honeybees (apis mellifera) and mites (varroa destructor). J. Gen. Virol. 2005, 86, 3419–3424. [Google Scholar] [CrossRef]
- Craggs, J.K.; Ball, J.K.; Thomson, B.J.; Irving, W.L.; Grabowska, A.M. Development of a strand-specific rt-pcr based assay to detect the replicative form of hepatitis c virus rna. J. Virol Methods 2001, 94, 111–120. [Google Scholar] [CrossRef]
- Guu, T.S.; Zheng, W.; Tao, Y.J. Bunyavirus: Structure and replication. In Viral Molecular Machines; Springer: Boston, MA, USA, 2012; pp. 245–266. [Google Scholar]
- De Miranda, J.R.; Bailey, L.; Ball, B.V.; Blanchard, P.; Budge, G.E.; Chejanovsky, N.; Chen, Y.-P.; Gauthier, L.; Genersch, E.; de Graaf, D.C.; et al. Standard methods for virus research in apis mellifera. J. Apic. Res. 2015, 52, 1–56. [Google Scholar] [CrossRef]
- R Core Team. Team. R: A language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. Available online: Https://www.R-project.Org (accessed on 2 September 2019).
- Stevenson, M.; Nunes, T.; Sanchez, J.; Thornton, R.; Reiczigel, J.; Robison-Cox, J.; Sebastiani, P. Epir: An r package for the analysis of epidemiological data. R package version 1.0-4, 2013. [Google Scholar]
- Felden, A.; Paris, C.; Chapple, D.G.; Suarez, A.V.; Tsutsui, N.D.; Lester, P.J.; Gruber, M.A. Native and introduced argentine ant populations are characterised by distinct transcriptomic signatures associated with behaviour and immunity. NeoBiota 2019, 49, 105–126. [Google Scholar] [CrossRef]
- Thompson, J.; Higgins, D.; Gibson, T. Clustalw: Improving the sensitivity of progressive multiple sequence through weighing, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. Mega x: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. Beast 2: A software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Rieux, A.; Balloux, F. Inferences from tip-calibrated phylogenies: A review and a practical guide. Mol. Ecol. 2016, 25, 1911–1924. [Google Scholar] [CrossRef]
- Drummond, A.J.; Pybus, O.G.; Rambaut, A.; Forsberg, R.; Rodrigo, A.G. Measurably evolving populations. Trends Ecol. Evol. 2003, 18, 481–488. [Google Scholar] [CrossRef]
- Parker, J.; Rambaut, A.; Pybus, O.G. Correlating viral phenotypes with phylogeny: Accounting for phylogenetic uncertainty. Infect. Genet. Evol. 2008, 8, 239–246. [Google Scholar] [CrossRef]
- Wang, T.H.; Donaldson, Y.K.; Brettle, R.P.; Bell, J.E.; Simmonds, P. Identification of shared populations of human immunodeficiency virus type 1 infecting microglia and tissue macrophages outside the central nervous system. J. Virol. 2001, 75, 11686–11699. [Google Scholar] [CrossRef]
- Boncristiani, H.F., Jr.; Di Prisco, G.; Pettis, J.S.; Hamilton, M.; Chen, Y.P. Molecular approaches to the analysis of deformed wing virus replication and pathogenesis in the honey bee, apis mellifera. Virol. J. 2009, 6, 221. [Google Scholar] [CrossRef] [PubMed]
- Bonning, B.C.; Miller, W.A. Dicistroviruses. Annu. Rev. Entomol. 2010, 55, 129–150. [Google Scholar] [CrossRef] [PubMed]
- Meeus, I.; de Miranda, J.R.; de Graaf, D.C.; Wackers, F.; Smagghe, G. Effect of oral infection with kashmir bee virus and israeli acute paralysis virus on bumblebee (bombus terrestris) reproductive success. J. Invertebr. Pathol. 2014, 121, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Benaets, K.; Geystelen, A.V.; Cardoen, D.; Smet, L.D.; Graaf, D.C.d.; Schoofs, L.; Larmuseau, M.H.D.; Brettell, L.E.; Martin, S.J.; Wenseleers, T. Covert deformed wing virus infections have long-term deleterious effects on honeybee foraging and survival. Proc. Biol. Sci. 2017, 284, 20162149. [Google Scholar] [CrossRef]
- Martin, S.J.; Brettell, L.E. Deformed wing virus in honeybees and other insects. Annu. Rev. Virol. 2019, 6, 49–69. [Google Scholar] [CrossRef]
- Remnant, E.J.; Shi, M.; Buchmann, G.; Blacquiere, T.; Holmes, E.C.; Beekman, M.; Ashe, A. A diverse range of novel rna viruses in geographically distinct honey bee populations. J. Virol. 2017, 91, e00158-17. [Google Scholar] [CrossRef]
- Seabloom, E.W.; Borer, E.T.; Gross, K.; Kendig, A.E.; Lacroix, C.; Mitchell, C.E.; Mordecai, E.A.; Power, A.G. The community ecology of pathogens: Coinfection, coexistence and community composition. Ecol. Lett. 2015, 18, 401–415. [Google Scholar] [CrossRef]
- Daszak, P.; Cunningham, A.A.; Hyatt, A.D. Anthropogenic environmental change and the emergence of infectious diseases in wildlife. Acta Trop. 2001, 78, 103–116. [Google Scholar] [CrossRef]
- Lembo, T.; Hampson, K.; Haydon, D.T.; Craft, M.; Dobson, A.; Dushoff, J.; Ernest, E.; Hoare, R.; Kaare, M.; Mlengeya, T.; et al. Exploring reservoir dynamics: A case study of rabies in the serengeti ecosystem. J. Appl. Ecol. 2008, 45, 1246–1257. [Google Scholar] [CrossRef]
- Wilfert, L.; Long, G.; Leggett, H.C.; Schmid-Hempel, P.; Butlin, R.; Martin, S.J.; Boots, M. Deformed wing virus is a recent global epidemic in honeybees driven by varroa mites. Science 2016, 351, 594–597. [Google Scholar] [CrossRef]
- Ravoet, J.; De Smet, L.; Meeus, I.; Smagghe, G.; Wenseleers, T.; de Graaf, D.C. Widespread occurrence of honey bee pathogens in solitary bees. J. Invertebr. Pathol. 2014, 122, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Manley, R.; Temperton, B.; Boots, M.; Wilfert, L. Contrasting impacts of a novel specialist vector on multihost viral pathogen epidemiology in wild and managed bees. Mol. Ecol. 2020, 29, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Dobson, A. Population dynamics of pathogens with multiple host species. Am. Nat. 2004, 164, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Alizon, S.; de Roode, J.C.; Michalakis, Y. Multiple infections and the evolution of virulence. Ecol. Lett. 2013, 16, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Telfer, S.; Lambin, X.; Birtles, R.; Beldomenico, P.; Burthe, S.; Paterson, S.; Begon, M. Species interactions in a parasite community drive infection risk in a wildlife population. Science 2010, 330, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Natsopoulou, M.E.; McMahon, D.P.; Doublet, V.; Frey, E.; Rosenkranz, P.; Paxton, R.J. The virulent, emerging genotype b of deformed wing virus is closely linked to overwinter honeybee worker loss. Sci. Rep. 2017, 7, 5242. [Google Scholar] [CrossRef]
- Mordecai, G.J.; Brettell, L.E.; Martin, S.J.; Dixon, D.; Jones, I.M.; Schroeder, D.C. Superinfection exclusion and the long-term survival of honey bees in varroa-infested colonies. ISME J. 2016, 10, 1182–1191. [Google Scholar] [CrossRef]
- Mideo, N. Parasite adaptations to within-host competition. Trends Parasitol. 2009, 25, 261–268. [Google Scholar] [CrossRef]
- Natsopoulou, M.E.; McMahon, D.P.; Doublet, V.; Bryden, J.; Paxton, R.J. Interspecific competition in honeybee intracellular gut parasites is asymmetric and favours the spread of an emerging infectious disease. Proc. Biol. Sci. 2015, 282, 20141896. [Google Scholar] [CrossRef]
- Lello, J.; Boag, B.; Fenton, A.; Stevenson, I.R.; Hudson, P.J. Competition and mutualism among the gut helminths of a mammalian host. Nature 2004, 428, 840–844. [Google Scholar] [CrossRef]
- Seppälä, O.; Karvonen, A.; Valtonen, E.T.; Jokela, J. Interactions among co-infecting parasite species: A mechanism maintaining genetic variation in parasites? Proc. Biol. Sci. 2009, 276, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Rigaud, T.; Perrot-Minnot, M.J.; Brown, M.J. Parasite and host assemblages: Embracing the reality will improve our knowledge of parasite transmission and virulence. Proc. Biol. Sci. 2010, 277, 3693–3702. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, S.D.; Ochoa, R.; Bauchan, G.; Gulbronson, C.; Mowery, J.D.; Cohen, A.; Lim, D.; Joklik, J.; Cicero, J.M.; Ellis, J.D.; et al. Varroa destructor feeds primarily on honey bee fat body tissue and not hemolymph. Proc. Natl. Acad. Sci. USA 2019, 116, 1792–1801. [Google Scholar]
- Gisder, S.; Aumeier, P.; Genersch, E. Deformed wing virus: Replication and viral load in mites (varroa destructor). J. Gen. Virol. 2009, 90, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Posada-Florez, F.; Childers, A.K.; Heerman, M.C.; Egekwu, N.I.; Cook, S.C.; Chen, Y.; Evans, J.D.; Ryabov, E.V. Deformed wing virus type a, a major honey bee pathogen, is vectored by the mite varroa destructor in a non-propagative manner. Sci. Rep. 2019, 9, 12445. [Google Scholar] [CrossRef] [PubMed]
- Porter, S.D.; Valles, S.M.; Pereira, R.M. Scavenging crickets (orthoptera: Gryllidae) transmit solenopsis invicta virus 3 to red imported fire ant (hymenoptera: Formicidae) colonies. Fla. Entomol. 2016, 99, 811–812. [Google Scholar] [CrossRef][Green Version]
- Le Sage, M.J.; Towey, B.D.; Brunner, J.L. Do scavengers prevent or promote disease transmission? The effect of invertebrate scavenging on ranavirus transmission. Funct. Ecol. 2019, 33, 1342–1350. [Google Scholar] [CrossRef]
- Mondet, F.; de Miranda, J.R.; Kretzschmar, A.; Le Conte, Y.; Mercer, A.R. On the front line: Quantitative virus dynamics in honeybee (apis mellifera l.) colonies along a new expansion front of the parasite varroa destructor. PLoS Pathog. 2014, 10, e1004323. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Family | Genus | Species | Common Name | n | DWV | KBV | MKV | BQCV | LhuBLV1 |
|---|---|---|---|---|---|---|---|---|---|---|
| Araneae | Salticidae | Helpis | Helpis minitabunda | jumping spider | 1 | + | - | + | - | + |
| Lycosidae | Lycosa | - | wolf spider | 1 | - | - | - | - | - | |
| Theridiidae | Steatoda | Steatoda capensis | black cobweb spider | 3 | - | + | +/+ | - | - | |
| Thomisidae | Diaea | flower spider | 4 | - | - | - | - | - | ||
| Blattodea | Blattidae | Celatoblatta | - | cockroach | 9 | +/+ | + | + | - | - |
| Maoriblatta | - | cockroach | 7 | + | - | + | - | + | ||
| Coleoptera | Curculionidae | Scolopterus | Scolopterus penicillatus | black spined weevil | 1 | + | - | - | - | - |
| Diptera | Sarcophagidae | Jantia | Jantia crassipalpis | flesh fly | 2 | - | - | - | - | - |
| Stratiomyidae | - | - | solider fly | 2 | - | - | - | - | + | |
| Tipulidae | Leptotarsus | - | crane fly | 2 | + | - | - | - | - | |
| Dermaptera | Forficulidae | Forficula | Forficula auricularia | European earwig | 2 | + | + | - | - | - |
| Hymenoptera | Apidae | Apis | Apis mellifera | honey bee | 51 | +/+ | +/+ | + | + | - |
| Bombus | - | bumble bee | 2 | - | +/+ | - | - | - | ||
| Formicidae | Linepithema | Linepithema humile | Argentine ant | 32 | +/+ | +/+ | +/+ | - | +/+ | |
| Pompilidae | Sphictostethus | Sphictostethus nitidus | golden hunter wasp | 1 | + | - | - | - | - | |
| Vespidae | Polistes | Polistes chinensis | Chinese paper wasp | 11 | + | - | +/+ | - | - | |
| Polistes humilis | Australian paper wasp | 2 | - | - | + | - | - | |||
| Vespula | Vespula germanica | German wasp | 1 | +/+ | + | +/+ | - | - | ||
| Vespula vulgaris | common wasp | 3 | +/+ | +/+ | +/+ | - | - | |||
| Ichneumonidae | Xanthocryptus | Xanthocryptus novozealandicus | lemon tree borer parasite | 1 | + | - | - | - | - | |
| Odonata | Lestidae | Austrolestes | Austrolestes colensonis | blue damselfly | 2 | - | - | - | - | - |
| Orthoptera | Acrididae | Locusta | Locusta migratoria | migratory locust | 1 | - | - | - | - | - |
| Gryllidae | Bobilla | - | small field cricket | 6 | + | +/+ | - | - | + | |
| Teleogryllus | Teleogryllus commodus | black field cricket | 4 | - | - | - | - | - | ||
| Total virus-positive species (samples) | 21 (151) | 14 (83) | 9 (44) | 10 (53) | 1 (18) | 5 (32) | ||||
| Host | BQCV | DWV | KBV | LhuBLV1 | MKV |
|---|---|---|---|---|---|
| A. mellifera | 35% (23%–49%) | 100%* (94%–100%) | 25% (15%–39%) | - | 41% (28%–55%) |
| L. humile | - | 41% (24%–58%) | 56%* (39%–72%) | 78% (61%–90%) | 34% (20%–53%) |
| Statistic | n | Observed to Expected Ratio (95% CI) | Observed Mean (95% CI) | Null Mean (95% CI) | p Value |
|---|---|---|---|---|---|
| Association Index (AI) | |||||
| Geographic location | - | 0.21 (0.13–0.30) | 2.83 (1.95–3.74) | 13.43 (12.47–14.47) | <0.01 |
| Host species | - | 0.55 (0.40–0.74) | 4.96 (3.89–6.01) | 8.98 (8.17–9.76) | <0.01 |
| Parsimony Score (PS) | |||||
| Geographic location | - | 0.35 (0.30–0.40) | 30.91 (28.00–34.00) | 88.56 (84.43–92.45) | <0.01 |
| Host species | - | 0.68 (0.61–0.74) | 34.87 (32.00–37.00) | 51.30 (49.75–52.35) | <0.01 |
| Maximum Clade (MC) scores | |||||
| Asia | 50 | - | 15.59 (11.00–21.00) | 2.57 (2.12–3.27) | <0.01 |
| Europe | 76 | - | 14.44 (14.00–16.00) | 3.53 (2.89–4.79) | <0.01 |
| North America | 24 | - | 4.48 (3.00–8.00) | 1.66 (1.28–2.09) | <0.01 |
| Oceania | 25 | - | 12.54 (6.00–18.00) | 1.72 (1.29–2.24) | <0.01 |
| Hawaii | 3 | - | 1.07 (1.00–2.00) | 1.01 (1.00–1.06) | 1 |
| Apis | 126 | - | 13.24 (9.00–21.00) | 7.04 (5.37–9.93) | 0.01 |
| Varroa destructor | 13 | - | 1.70 (1.00–3.00) | 1.27 (1.01–1.99) | 0.04 |
| Associate | 6 | - | 1.83 (1.00–3.00) | 1.05 (1.00–1.19) | <0.01 |
| Non-Apis bee | 23 | - | 4.23 (4.00–5.00) | 1.63 (1.23–2.07) | <0.01 |
| Vespidae | 5 | - | 1.09 (1.00–2.00) | 1.03 (1.00–1.15) | 1 |
| Formicidae | 4 | - | 2.00 (2.00–2.00) | 1.02 (1.00–1.07) | <0.01 |
| Other Hymenoptera | 2 | - | 1.00 (1.00–1.00) | 1.00 (1.00–1.00) | 1 |
| Statistic | n | Observed to Expected Ratio (95% CI) | Observed Mean (95%CI) | Null Mean (95%CI) | p Value |
|---|---|---|---|---|---|
| Association Index (AI) | |||||
| Geographic location | 0.16 (0.13–0.25) | 0.36 (0.33–0.43) | 2.19 (1.74–2.63) | <0.01 | |
| Host species | 0.27 (0.11–0.50) | 0.58 (0.29–0.79) | 2.14 (1.59–2.57) | <0.01 | |
| Parsimony Score (PS) | |||||
| Geographic location | 0.45 (0.40–0.58) | 6.06 (6.00–7.00) | 13.44 (12.12–14.92) | <0.01 | |
| Host species | 0.59 (0.50–0.73) | 7.55 (7.00–8.00) | 12.74 (11.00–13.98) | <0.01 | |
| Maximum Clade (MC) scores | |||||
| North America | 4 | - | 2.00 (2.00–2.00) | 1.26 (1.00–2.00) | 0.10 |
| New Zealand | 9 | - | 7.83 (6.00–7.00) | 1.92 (1.07–3.00) | 0.01 |
| Europe | 3 | - | 3.00 (3.00–3.00) | 1.10 (1.00–2.00) | 0.01 |
| Australia | 5 | - | 4.37 (4.00–5.00) | 1.24 (1.00–2.00) | 0.01 |
| Apis | 6 | - | 2.54 (2.00–4.00) | 1.45 (1.00–2.94) | 0.19 |
| Bombus | 3 | - | 2.97 (2.00–3.00) | 1.10 (1.00–2.00) | <0.01 |
| Linepithema | 10 | - | 3.08 (3.00–4.00) | 2.12 (1.10–3.44) | 0.16 |
| associate | 2 | - | 1.09 (1.00–2.00) | 1.03 (1.00–1.17) | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobelmann, J.; Felden, A.; Lester, P.J. Genetic Strain Diversity of Multi-Host RNA Viruses that Infect a Wide Range of Pollinators and Associates is Shaped by Geographic Origins. Viruses 2020, 12, 358. https://doi.org/10.3390/v12030358
Dobelmann J, Felden A, Lester PJ. Genetic Strain Diversity of Multi-Host RNA Viruses that Infect a Wide Range of Pollinators and Associates is Shaped by Geographic Origins. Viruses. 2020; 12(3):358. https://doi.org/10.3390/v12030358
Chicago/Turabian StyleDobelmann, Jana, Antoine Felden, and Philip J. Lester. 2020. "Genetic Strain Diversity of Multi-Host RNA Viruses that Infect a Wide Range of Pollinators and Associates is Shaped by Geographic Origins" Viruses 12, no. 3: 358. https://doi.org/10.3390/v12030358
APA StyleDobelmann, J., Felden, A., & Lester, P. J. (2020). Genetic Strain Diversity of Multi-Host RNA Viruses that Infect a Wide Range of Pollinators and Associates is Shaped by Geographic Origins. Viruses, 12(3), 358. https://doi.org/10.3390/v12030358