Phylogenetic and Demographic Characterization of Directed HIV-1 Transmission Using Deep Sequences from High-Risk and General Population Cohorts/Groups in Uganda

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
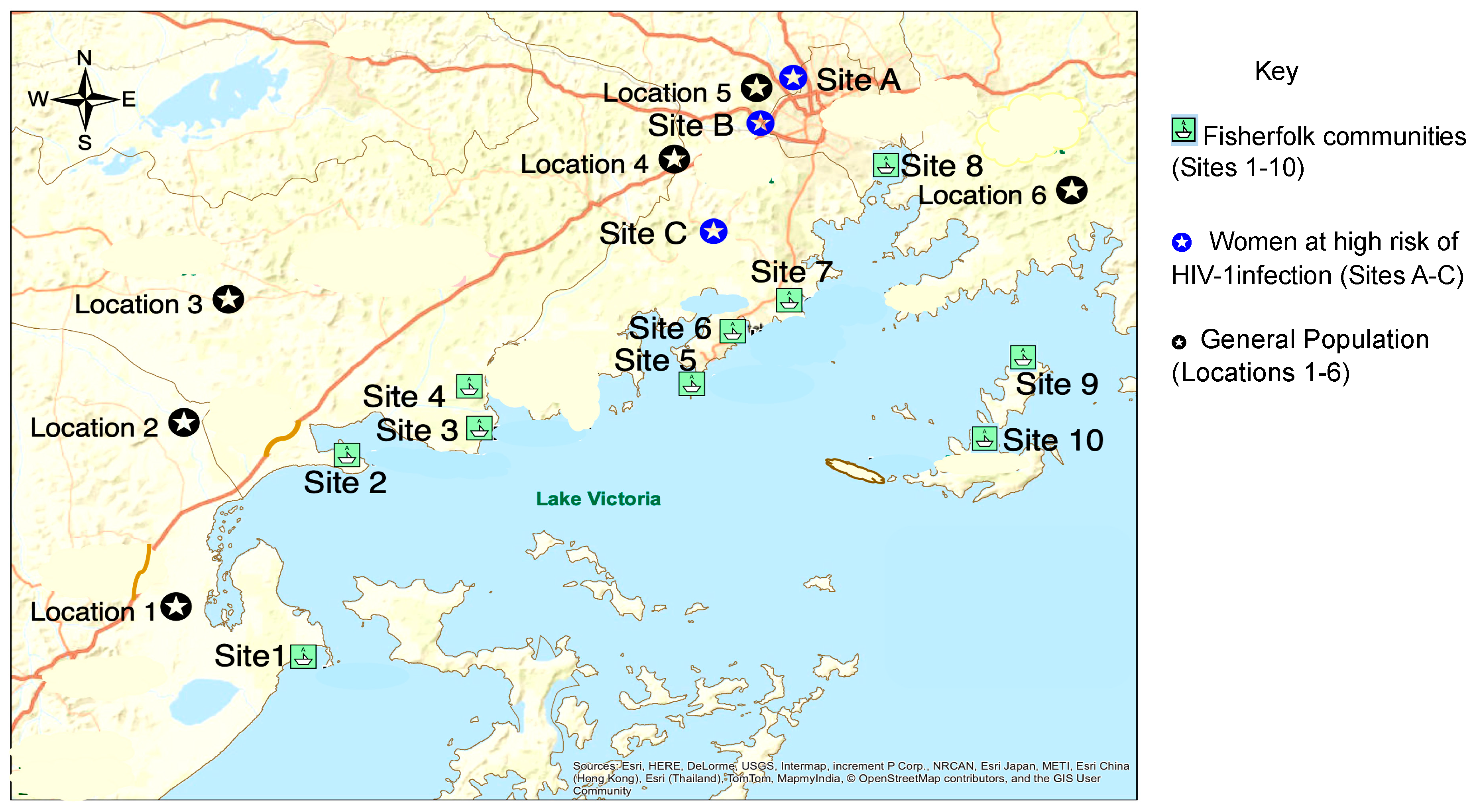
2.1. Study Design and Population
2.2. Ethics Statement
2.3. Deep Sequencing and Assembly of HIV-1 Reads
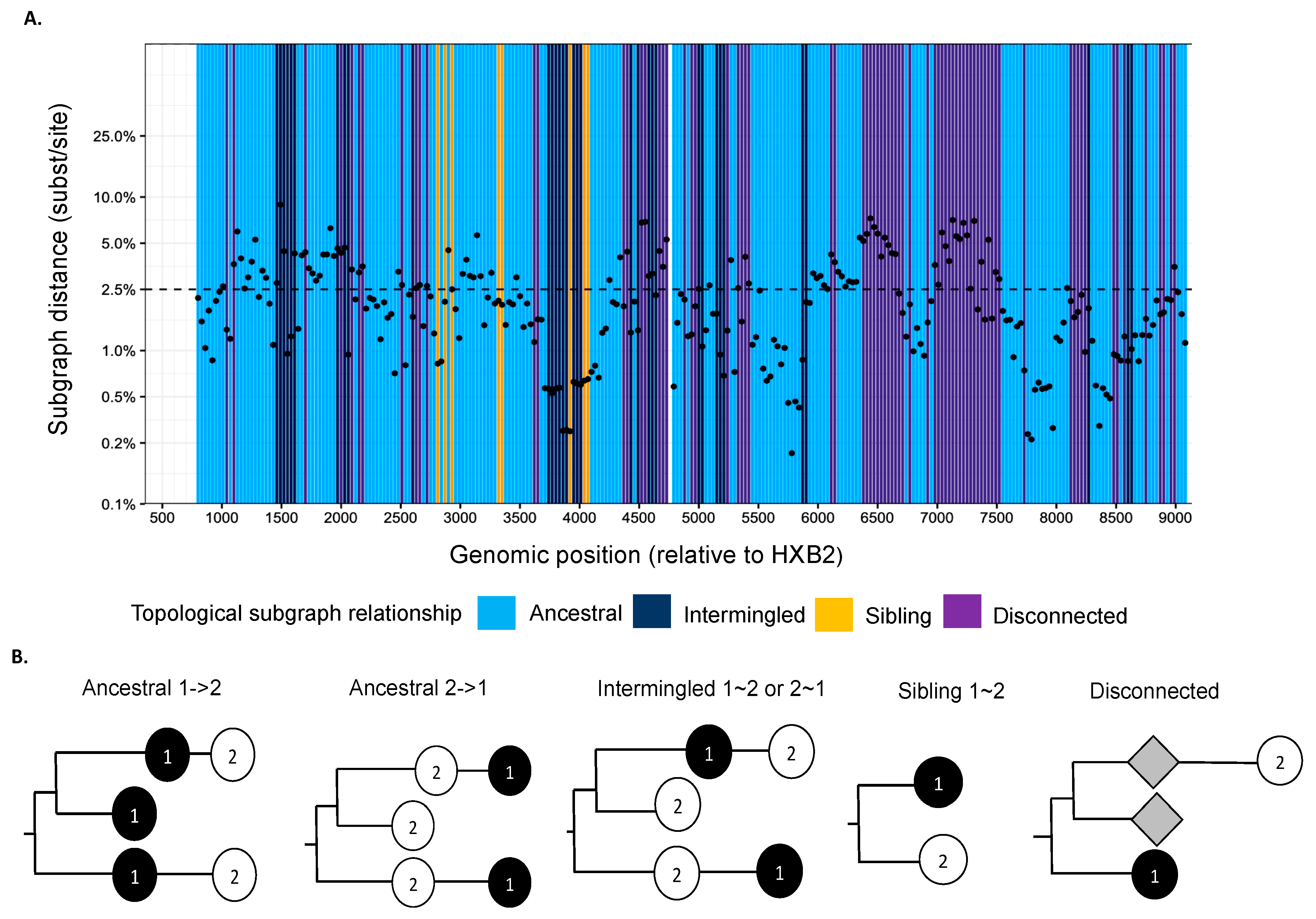
2.4. Deep-Sequence Phylogenetic Analysis
2.5. Reconstruction of Transmission Networks
2.6. Quantifying HIV-1 Transmission Flows
3. Results
3.1. Population-Based Sample of HIV-1 Deep Sequences
3.2. Reconstructed HIV-1 Transmission Networks
3.3. HIV-1 Transmission within and between Study Participants of the General Population, Fisherfolk, and the Women at High Risk Cohort and their Clients
3.4. HIV Transmission by Gender
3.5. HIV-1 Transmission by Age Groups
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Name | Institution | Representatives & Roles | Email Address |
|---|---|---|---|
| Lucie Abeler-Dörner | University of Oxford | Project manager | lucie.abeler-dorner@bdi.ox.ac.uk |
| Helen Ayles | PopART/Zambart | PopART | helen@zambart.org.zm |
| David Bonsall | University of Oxford | Sequencing lab | david.bonsall@bdi.ox.ac.uk |
| Rory Bowden | University of Oxford | Sequencing lab | rbowden@well.ox.ac.uk |
| Vincent Calvez | Institut Pasteur | TasP trial | vincent.calvez@me.com |
| Max Essex | Harvard Botswana | Botswana studies | messex@hsph.harvard.edu |
| Sarah Fidler | PopART/Imperial College London | PopART | s.fidler@imperial.ac.uk |
| Christophe Fraser | University of Oxford | Principal Investigator PANGEA 2, Executive Committee and PopART Phylogenetics | christophe.fraser@bdi.ox.ac.uk |
| Kate Grabowski | Johns Hopkins University | Executive Committee and Rakai | mgrabows@jhu.edu |
| Tanya Golubchik | University of Oxford | Data manager | tanya.golubchik@bdi.ox.ac.uk |
| Richard Hayes | PopART/LSHTM | PopART | Richard.Hayes@lshtm.ac.uk |
| Joshua Herbeck | University of Washington | Partners PrEP; Partners in Prevention | herbeck@uw.edu |
| Joseph Kagaayi | Rakai Health Sciences Program | Rakai Health Sciences Program | jkagayi@rhsp.org |
| Pontiano Kaleebu | MRC/UVRI Uganda | MRC studies | pontiano.kaleebu@mrcuganda.org |
| Jairam Lingappa | University of Washington | Partners PrEP; Partners in Prevention | lingappa@uw.edu |
| Sikhulile Moyo | Botswana Harvard AIDS Institute Partnership | Botswana studies | sikhulilemoyo@gmail.com |
| Vladimir Novitsky | Harvard University | Botswana studies | vnovi@hsph.harvard.edu |
| Deenan Pillay | Africa Health Research Institute (AHRI)/University College London | Principal Investigator PANGEA-1/Executive Committee, AHRI studies | dpillay@ahri.org |
| Thomas Quinn | Johns Hopkins University | Rakai Health Sciences Program | tquinn2@jhmi.edu |
| Andrew Rambaut | University of Edinburgh | Executive Committee | a.rambaut@ed.ac.uk |
| Oliver Ratmann | Imperial College London | Analysis | oliver.ratmann@imperial.ac.uk |
| Janet Seeley | MRC/UVRI Uganda/LSHTM | MRC Uganda | Janet.Seeley@LSHTM.ac.uk |
| Deogratius Ssemwanga | MRC/UVRI Uganda | MRC studies | Deogratius.Ssemwanga@mrcuganda.org |
| Frank Tanser | Africa Health Research Institute | AHRI studies | ftanser@lincoln.ac.uk |
| Maria Wawer | Johns Hopkins University | Rakai Health Sciences Program | mwawer1@jhu.edu |
Appendix B
| Name | Institution | Study Team Representatives | Email Address |
|---|---|---|---|
| Myron Cohen | University of North Carolina | -------- | myron_cohen@med.unc.edu |
| Tulio D’Oliveira | University of KwaZulu-Natal | -------- | tuliodna@gmail.com |
| Ann Dennis | University of North Carolina | -------- | ann_dennis@med.unc.edu |
| Max Essex | Harvard Botswana | Botswana studies | messex@hsph.harvard.edu |
| Sarah Fidler | PopART/Imperial College London | PopART Phylogenetics | s.fidler@imperial.ac.uk |
| Dan Frampton | University College London | -------- | d.frampton@ucl.ac.uk |
| Christophe Fraser | University of Oxford | PopART Phylogenetics | christophe.fraser@well.ox.ac.uk |
| Tanya Golubchik | University of Oxford | tanya.golubchik@bdi.ox.ac.uk | |
| Richard Hayes | PopART/LSHTM | PopART Phylogenetics | Richard.Hayes@lshtm.ac.uk |
| Josh Herbeck | University of Washington | Partners PrEP; Partners in Prevention | herbeck@uw.edu |
| Anne Hoppe | University College London | Project Manager PANGEA 1/EARNEST | a.hoppe@ucl.ac.uk; hoppe.anne@gmail.com |
| Pontiano Kaleebu | MRC/UVRI Uganda | MRC studies | pontiano.kaleebu@mrcuganda.org |
| Paul Kellam | Cambridge University | -------- | paul.kellam@kymab.com |
| Cissy Kityo | EARNEST/JCRC Uganda | EARNEST | ckityo@jcrc.org.ug |
| Andrew Leigh-Brown | University of Edinburgh | -------- | A.Leigh-Brown@ed.ac.uk |
| Jairam Lingappa | University of Washington | Partners PrEP; Partners in Prevention | lingappa@uw.edu |
| Vladimir Novitsky | Harvard University | Botswana studies | vnovi@hsph.harvard.edu |
| Nick Paton | EARNEST/University of Singapore | EARNEST | nick.paton@ucl.ac.uk |
| Deenan Pillay | Africa Health Research Institute/University College London | Principal Investigator PANGEA 1/Africa Health Research Institute studies | dpillay@ahri.org |
| Tom Quinn | Johns Hopkins University | Rakai Health Sciences Program | tquinn2@jhmi.edu |
| Oliver Ratmann | Imperial College London | -------- | oliver.ratmann@imperial.ac.uk |
| Deogratius Ssemwanga | MRC/UVRI Uganda | MRC studies | Deogratius.Ssemwanga@mrcuganda.org |
| Frank Tanser | Africa Health Research Institute | -------- | ftanser@gmail.com |
| Maria Wawer | Johns Hopkins University | Rakai Health Sciences Program | mwawer1@jhu.edu |
References
- UNAIDS DATA 2019. Available online: https://www.unaids.org/sites/default/files/media_asset/2019-UNAIDS-data_en.pdf (accessed on 19 December 2019).
- Uganda AIDS Commision. Uganda HIV/AIDS Country Progress Report July 2017–June 2018; Uganda AIDS Commission: Kampala, Uganda, 2018; p. 91.
- Ministry of Health and Uganda AIDS Commission. Multi-Sectoral HIV Programming for MARPS in Uganda: Review of Profiles, Sizes and Programme Coverage; Review Report; Ministry of Health and Uganda AIDS Commission: Kampala, Uganda, 2014.
- Ministry of Health Uganda. Population-Based HIV Impact Assessement (UPHIA) 2016–2017. Summary Sheet: Preliminary Findings; Ministry of Health Uganda: Kampala, Uganda, 2017.
- Kamali, A.; Nsubuga, R.N.; Ruzagira, E.; Bahemuka, U.; Asiki, G.; Price, M.A.; Newton, R.; Kaleebu, P.; Fast, P. Heterogeneity of HIV incidence: A comparative analysis between fishing communities and in a neighbouring rural general population, Uganda, and implications for HIV control. Sex. Transm. Infect. 2016, 92, 447–454. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kasamba, I.; Nash, S.; Seeley, J.; Weiss, H.A. HIV incidence among women at high risk of HIV infection attending a dedicated clinic in Kampala, Uganda: 2008–2017. Sex. Transm. Dis. 2019, 46, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Wabwire-Mangen, F.; Odiit, M.; Kirungi, W.; Kisitu, D.K.; Wanyama, J.O. HIV Modes of Transmission and Prevention Response Analysis; Uganda National AIDS Commission: Kampala, Uganda, 2009.
- UNAIDS Fast-Track—Ending the AIDS Epidemic by 2030. Available online: http://www.unaids.org/en/resources/documents/2014/JC2686_WAD2014report (accessed on 16 February 2018).
- UNAIDS/WHO Working Group on Global HIV/AIDS and STI Surveillance; Joint United Nations Programme on HIV/AIDS; World Health Organization. Guidelines for Second Generation HIV Surveillance: An Update: Know Your Epidemic; WHO Press: Geneva, Switzerland, 2013; ISBN 978-92-4-150582-6. [Google Scholar]
- Cuadros, D.F.; Li, J.; Branscum, A.J.; Akullian, A.; Jia, P.; Mziray, E.N.; Tanser, F. Mapping the spatial variability of HIV infection in Sub-Saharan Africa: Effective information for localized HIV prevention and control. Sci. Rep. 2017, 7, 9093. [Google Scholar] [CrossRef] [PubMed]
- Barankanira, E.; Molinari, N.; Niyongabo, T.; Laurent, C. Spatial analysis of HIV infection and associated individual characteristics in Burundi: Indications for effective prevention. BMC Public Health 2016, 16, 118. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.-J.; Cherutich, P.; Kilonzo, N.; Cremin, I.; Fecht, D.; Kimanga, D.; Harper, M.; Masha, R.L.; Ngongo, P.B.; Maina, W.; et al. Maximising the effect of combination HIV prevention through prioritisation of the people and places in greatest need: A modelling study. Lancet 2014, 384, 249–256. [Google Scholar] [CrossRef]
- Kuteesa, M.O.; Weiss, H.A.; Abaasa, A.; Nash, S.; Nsubuga, R.N.; Newton, R.; Seeley, J.; Kamali, A. Feasibility of conducting HIV combination prevention interventions in fishing communities in Uganda: A pilot cluster randomised trial. PLoS ONE 2019, 14, e0210719. [Google Scholar] [CrossRef]
- Kagaayi, J.; Chang, L.W.; Ssempijja, V.; Grabowski, M.K.; Ssekubugu, R.; Nakigozi, G.; Kigozi, G.; Serwadda, D.M.; Gray, R.H.; Nalugoda, F.; et al. Impact of combination HIV interventions on HIV incidence in hyperendemic fishing communities in Uganda: A prospective cohort study. Lancet HIV 2019, 6, e680–e687. [Google Scholar] [CrossRef]
- Ssewamala, F.M.; Sensoy Bahar, O.; Tozan, Y.; Nabunya, P.; Mayo-Wilson, L.J.; Kiyingi, J.; Kagaayi, J.; Bellamy, S.; McKay, M.M.; Witte, S.S. A combination intervention addressing sexual risk-taking behaviors among vulnerable women in Uganda: Study protocol for a cluster randomized clinical trial. BMC Women’s Health 2019, 19, 111. [Google Scholar] [CrossRef]
- Rambaut, A.; Robertson, D.L.; Pybus, O.G.; Peeters, M.; Holmes, E.C. Human immunodeficiency virus. Phylogeny and the origin of HIV-1. Nature 2001, 410, 1047–1048. [Google Scholar] [CrossRef]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef]
- Faria, N.R.; Rambaut, A.; Suchard, M.A.; Baele, G.; Bedford, T.; Ward, M.J.; Tatem, A.J.; Sousa, J.D.; Arinaminpathy, N.; Pépin, J.; et al. The early spread and epidemic ignition of HIV-1 in human populations. Science 2014, 346, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Kiwuwa-Muyingo, S.; Nazziwa, J.; Ssemwanga, D.; Ilmonen, P.; Njai, H.; Ndembi, N.; Parry, C.; Kitandwe, P.K.; Gershim, A.; Mpendo, J.; et al. HIV-1 transmission networks in high risk fishing communities on the shores of Lake Victoria in Uganda: A phylogenetic and epidemiological approach. PLoS ONE 2017, 12, e0185818. [Google Scholar] [CrossRef] [PubMed]
- Aldous, J.L.; Pond, S.K.; Poon, A.; Jain, S.; Qin, H.; Kahn, J.S.; Kitahata, M.; Rodriguez, B.; Dennis, A.M.; Boswell, S.L.; et al. Characterizing HIV Transmission Networks Across the United States. Clin. Infect. Dis. 2012, 55, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Leigh Brown, A.J.; Lycett, S.J.; Weinert, L.; Hughes, G.J.; Fearnhill, E.; Dunn, D.T. UK HIV drug resistance collaboration transmission network parameters estimated from HIV sequences for a nationwide epidemic. J. Infect. Dis. 2011, 204, 1463–1469. [Google Scholar] [CrossRef]
- Lewis, F.; Hughes, G.J.; Rambaut, A.; Pozniak, A.; Leigh Brown, A.J. Episodic Sexual Transmission of HIV revealed by molecular phylodynamics. PLoS Med. 2008, 5, e50. [Google Scholar] [CrossRef]
- Yebra, G.; Ragonnet-Cronin, M.; Ssemwanga, D.; Parry, C.M.; Logue, C.H.; Cane, P.A.; Kaleebu, P.; Brown, A.J.L. Analysis of the history and spread of HIV-1 in Uganda using phylodynamics. J. Gen. Virol. 2015, 96, 1890–1898. [Google Scholar] [CrossRef]
- Gray, R.R.; Tatem, A.J.; Lamers, S.; Hou, W.; Laeyendecker, O.; Serwadda, D.; Sewankambo, N.; Gray, R.H.; Wawer, M.; Quinn, T.C.; et al. Spatial phylodynamics of HIV-1 epidemic emergence in east Africa. AIDS 2009, 23, F9–F17. [Google Scholar] [CrossRef]
- de Oliveira, T.; Kharsany, A.B.; Gräf, T.; Cawood, C.; Khanyile, D.; Grobler, A.; Puren, A.; Madurai, S.; Baxter, C.; Karim, Q.A.; et al. Transmission networks and risk of HIV infection in KwaZulu-Natal, South Africa: A community-wide phylogenetic study. Lancet HIV 2017, 4, e41–e50. [Google Scholar] [CrossRef]
- Coltart, C.; Tanser, F.; Gareta, D.; Ntuli, S.; de Oliveira, T.; Pillay, D.; Johnson, A.; Hue, S. The identification of a micro-epidemic in a hyper-endemic HIV setting using molecular epidemiology. J. Int. AIDS Soc. Mexico 2018, 21, e25148. [Google Scholar]
- Rasmussen, D.A.; Wilkinson, E.; Vandormael, A.; Tanser, F.; Pillay, D.; Stadler, T.; de Oliveira, T. Tracking external introductions of HIV using phylodynamics reveals a major source of infections in rural KwaZulu-Natal, South Africa. Virus Evol. 2018, 4, vey037. [Google Scholar] [CrossRef]
- Grabowski, M.K.; Lessler, J.; Redd, A.D.; Kagaayi, J.; Laeyendecker, O.; Ndyanabo, A.; Nelson, M.I.; Cummings, D.A.T.; Bwanika, J.B.; Mueller, A.C.; et al. The role of viral introductions in sustaining community-based HIV epidemics in rural Uganda: Evidence from spatial clustering, phylogenetics, and egocentric transmission models. PLoS Med. 2014, 11, e1001610. [Google Scholar] [CrossRef] [PubMed]
- Bbosa, N.; Ssemwanga, D.; Nsubuga, R.N.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Nanyonjo, M.; Kuteesa, M.; Seeley, J.; Kiwanuka, N.; Bagaya, B.S.; et al. Phylogeography of HIV-1 suggests that Ugandan fishing communities are a sink for, not a source of, virus from general populations. Sci. Rep. 2019, 9, 1051. [Google Scholar] [CrossRef] [PubMed]
- Ratmann, O.; Kagaayi, J.; Hall, M.; Golubchick, T.; Kigozi, G.; Xi, X.; Wymant, C.; Nakigozi, G.; Abeler-Dörner, L.; Bonsall, D.; et al. Quantifying HIV transmission flow between high-prevalence hotspots and surrounding communities: A population-based study in Rakai, Uganda. Lancet HIV 2020, 7, e173–e183. [Google Scholar] [CrossRef]
- Abeler-Dörner, L.; Grabowski, M.K.; Rambaut, A.; Pillay, D.; Fraser, C. PANGEA consortium PANGEA-HIV 2: Phylogenetics and networks for generalised epidemics in Africa. Curr. Opin. HIV AIDS 2019, 14, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Asiki, G.; Mpendo, J.; Abaasa, A.; Agaba, C.; Nanvubya, A.; Nielsen, L.; Seeley, J.; Kaleebu, P.; Grosskurth, H.; Kamali, A. HIV and syphilis prevalence and associated risk factors among fishing communities of Lake Victoria, Uganda. Sex. Transm. Infect. 2011, 87, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Asiki, G.; Murphy, G.; Nakiyingi-Miiro, J.; Seeley, J.; Nsubuga, R.N.; Karabarinde, A.; Waswa, L.; Biraro, S.; Kasamba, I.; Pomilla, C.; et al. The general population cohort in rural south-western Uganda: A platform for communicable and non-communicable disease studies. Int. J. Epidemiol. 2013, 42, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Omooja, J.; Nannyonjo, M.; Sanyu, G.; Nabirye, S.E.; Nassolo, F.; Lunkuse, S.; Kapaata, A.; Segujja, F.; Kateete, D.P.; Ssebaggala, E.; et al. Rates of HIV-1 virological suppression and patterns of acquired drug resistance among fisherfolk on first-line antiretroviral therapy in Uganda. J. Antimicrob. Chemother. 2019, 74, 3021–3029. [Google Scholar] [CrossRef]
- Salome, T.; Kasamba, I.; Mayanja, B.N.; Kazooba, P.; Were, J.; Kaleebu, P.; Munderi, P. The effect of Tenofovir on renal function among Ugandan adults on long-term antiretroviral therapy: A cross-sectional enrolment analysis. AIDS Res. Ther. 2016, 13, 28. [Google Scholar] [CrossRef]
- Namale, G.; Kamacooko, O.; Bagiire, D.; Mayanja, Y.; Abaasa, A.; Kilembe, W.; Price, M.; Ssemwanga, D.; Lunkuse, S.; Nanyonjo, M.; et al. Sustained virological response and drug resistance among female sex workers living with HIV on antiretroviral therapy in Kampala, Uganda: A cross-sectional study. Sex. Transm. Infect. 2019, 95, 405–411. [Google Scholar] [CrossRef]
- Nampijja, M.; Webb, E.L.; Kaweesa, J.; Kizindo, R.; Namutebi, M.; Nakazibwe, E.; Oduru, G.; Kabuubi, P.; Kabagenyi, J.; Kizito, D.; et al. The lake victoria island intervention study on worms and allergy-related diseases (LaVIISWA): Study protocol for a randomised controlled trial. Trials 2015, 16, 187. [Google Scholar] [CrossRef]
- Sanya, R.E.; Nkurunungi, G.; Hoek Spaans, R.; Nampijja, M.; O’Hara, G.; Kizindo, R.; Oduru, G.; Kabuubi Nakawungu, P.; Niwagaba, E.; Abayo, E.; et al. The impact of intensive versus standard anthelminthic treatment on allergy-related outcomes, helminth infection intensity and helminth-related morbidity in Lake Victoria fishing communities, Uganda: Results from the LaVIISWA cluster randomised trial. Clin. Infect. Dis. 2018, 68, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Abaasa, A.; Asiki, G.; Obuku Ekii, A.; Wanyenze, J.; Pala, P.; van Dam, G.J.; Corstjens, P.L.A.M.; Hughes, P.; Ding, S.; Pantaleo, G.; et al. Effect of high-intensity versus low-intensity praziquantel treatment on HIV disease progression in HIV and Schistosoma mansoni co-infected patients: A randomised controlled trial. Wellcome Open Res. 2018, 3, 81. [Google Scholar] [CrossRef] [PubMed]
- Vandepitte, J.; Bukenya, J.; Weiss, H.A.; Nakubulwa, S.; Francis, S.C.; Hughes, P.; Hayes, R.; Grosskurth, H. HIV and other sexually transmitted infections in a cohort of women involved in high-risk sexual behavior in Kampala, Uganda. Sex. Transm. Dis. 2011, 38, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Kapaata, A.; Lyagoba, F.; Ssemwanga, D.; Magambo, B.; Nanyonjo, M.; Levin, J.; Mayanja, B.N.; Mugasa, C.; Parry, C.M.; Kaleebu, P. HIV-1 subtype distribution trends and evidence of transmission clusters among incident cases in a rural clinical cohort in southwest Uganda, 2004–2010. AIDS Res. Hum. Retrovir. 2013, 29, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Abaasa, A.; Mbonye, M.; Asiki, G.; Ruzagira, E.; Price, M.; Fast, P.; Priddy, F.; Kaleebu, P.; Kamaali, A. Use of Fingerprinting Technology in HIV Prevention Studies. Experience from Fishing Communities in South-Western Uganda; HIV Research for Prevention (HIVR4P) conference; Poster Session: Chicago, IL, USA, 2016. [Google Scholar]
- Gall, A.; Morris, C.; Kellam, P.; Berry, N. Complete genome sequence of the WHO international standard for HIV-1 RNA determined by deep sequencing. Genome Announc. 2014, 2, e01254-13. [Google Scholar] [CrossRef] [PubMed]
- Yebra, G.; Frampton, D.; Gallo Cassarino, T.; Raffle, J.; Hubb, J.; Ferns, R.B.; Waters, L.; Tong, C.Y.W.; Kozlakidis, Z.; Hayward, A.; et al. A high HIV-1 strain variability in London, UK, revealed by full-genome analysis: Results from the ICONIC project. PLoS ONE 2018, 13, e0192081. [Google Scholar] [CrossRef]
- Wymant, C.; Blanquart, F.; Golubchik, T.; Gall, A.; Bakker, M.; Bezemer, D.; Croucher, N.J.; Hall, M.; Hillebregt, M.; Ong, S.H.; et al. Easy and accurate reconstruction of whole HIV genomes from short-read sequence data with shiver. Virus Evol. 2018, 4, vey007. [Google Scholar] [CrossRef]
- Wymant, C.; Hall, M.; Ratmann, O.; Bonsall, D.; Golubchik, T.; de Cesare, M.; Gall, A.; Cornelissen, M.; Fraser, C. STOP-HCV Consortium, The maela pneumococcal collaboration, and the BEEHIVE collaboration PHYLOSCANNER: Inferring transmission from within- and between-host pathogen genetic diversity. Mol. Biol. Evol. 2018, 35, 719–733. [Google Scholar] [CrossRef]
- Ratmann, O.; Grabowski, M.K.; Hall, M.; Golubchik, T.; Wymant, C.; Abeler-Dörner, L.; Bonsall, D.; Hoppe, A.; Brown, A.L.; de Oliveira, T.; et al. Inferring HIV-1 transmission networks and sources of epidemic spread in Africa with deep-sequence phylogenetic analysis. Nat. Commun. 2019, 10, 1411. [Google Scholar] [CrossRef]
- Romero-Severson, E.O.; Bulla, I.; Leitner, T. Phylogenetically resolving epidemiologic linkage. Proc. Natl. Acad. Sci. USA 2016, 113, 2690–2695. [Google Scholar] [CrossRef]
- Campbell, E.M.; Jia, H.; Shankar, A.; Hanson, D.; Luo, W.; Masciotra, S.; Owen, S.M.; Oster, A.M.; Galang, R.R.; Spiller, M.W.; et al. Detailed transmission network analysis of a large opiate-driven outbreak of HIV infection in the United States. J. Infect. Dis. 2017, 216, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, S.R.; Wertheim, J.O.; Bull, R.A.; Matthews, G.V.; Lamoury, F.M.J.; Scheffler, K.; Hellard, M.; Maher, L.; Dore, G.J.; Lloyd, A.R.; et al. A molecular transmission network of recent hepatitis C infection in people with and without HIV: Implications for targeted treatment strategies. J. Viral Hepat. 2017, 24, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Phyloscanner. Available online: https://github.com/BDI-pathogens/phyloscanner/tree/master/phyloflows (accessed on 21 December 2019).
- Gelman, A.; Rubin, D.B. Markov chain Monte Carlo methods in biostatistics. Stat. Methods Med. Res. 1996, 5, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Nazziwa, J.; Njai, H.F.; Ndembi, N.; Birungi, J.; Lyagoba, F.; Gershim, A.; Nakiyingi-Miiro, J.; Nielsen, L.; Mpendo, J.; Nanvubya, A.; et al. Short communication: HIV type 1 transmitted drug resistance and evidence of transmission clusters among recently infected antiretroviral-naive individuals from Ugandan fishing communities of Lake Victoria. AIDS Res. Hum. Retrovir. 2013, 29, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Alves, B.M.; Siqueira, J.D.; Prellwitz, I.M.; Botelho, O.M.; Da Hora, V.P.; Sanabani, S.; Recordon-Pinson, P.; Fleury, H.; Soares, E.A.; Soares, M.A. Estimating HIV-1 genetic diversity in Brazil through next-generation sequencing. Front. Microbiol. 2019, 10, 749. [Google Scholar] [CrossRef]
- Iwase, S.C.; Miyazato, P.; Katsuya, H.; Islam, S.; Yang, B.T.J.; Ito, J.; Matsuo, M.; Takeuchi, H.; Ishida, T.; Matsuda, K.; et al. HIV-1 DNA-capture-seq is a useful tool for the comprehensive characterization of HIV-1 provirus. Sci. Rep. 2019, 9, 12326. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Weaver, S.; Leigh Brown, A.J.; Wertheim, J.O. HIV-TRACE (TRAnsmission cluster engine): A tool for large scale molecular epidemiology of HIV-1 and other rapidly evolving pathogens. Mol. Biol. Evol. 2018, 35, 1812–1819. [Google Scholar] [CrossRef]
- Ragonnet-Cronin, M.; Hodcroft, E.; Hué, S.; Fearnhill, E.; Delpech, V.; Brown, A.J.L.; Lycett, S. UK HIV drug resistance database automated analysis of phylogenetic clusters. BMC Bioinform. 2013, 14, 317. [Google Scholar] [CrossRef]
- Gibson, K.M.; Jair, K.; Castel, A.D.; Bendall, M.L.; Wilbourn, B.; Jordan, J.A.; Crandall, K.A.; Pérez-Losada, M. DC Cohort Executive Committee A cross-sectional study to characterize local HIV-1 dynamics in Washington, DC using next-generation sequencing. Sci. Rep. 2020, 10, 1989. [Google Scholar] [CrossRef]
- Hauser, A.; Hofmann, A.; Meixenberger, K.; Altmann, B.; Hanke, K.; Bremer, V.; Bartmeyer, B.; Bannert, N. Increasing proportions of HIV-1 non-B subtypes and of NNRTI resistance between 2013 and 2016 in Germany: Results from the national molecular surveillance of new HIV-diagnoses. PLoS ONE 2018, 13, e0206234. [Google Scholar] [CrossRef]
- Skums, P.; Zelikovsky, A.; Singh, R.; Gussler, W.; Dimitrova, Z.; Knyazev, S.; Mandric, I.; Ramachandran, S.; Campo, D.; Jha, D.; et al. QUENTIN: Reconstruction of disease transmissions from viral quasispecies genomic data. Bioinformatics 2018, 34, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Giroir, B.P. The time is now to end the hiv epidemic. Am. J. Public Health 2020, 110, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Fauci, A.S.; Redfield, R.R.; Sigounas, G.; Weahkee, M.D.; Giroir, B.P. Ending the HIV epidemic: A plan for the United States. JAMA 2019, 321, 844. [Google Scholar] [CrossRef] [PubMed]



| Population and Gender | HIV-Positive Participants | Deep Sequenced | Proportion of HIV-Positive Participants Who Were Sequenced |
|---|---|---|---|
| Total | 6185 | 2531 | 40.9% |
| Fisherfolk | 2185 | 895 | 41.0% |
| Men | 1103 (50.5%) | 468 (52.3%) | 42.5% |
| Women | 1082 (49.5%) | 427 (47.7%) | 39.5% |
| General population | 3200 | 1309 | 40.9% |
| Men | 1578 (49.3%) | 636 (48.6%) | 40.3% |
| women | 1622 (50.7%) | 673 (51.4%) | 41.5% |
| Women at high risk and male clients | 800 | 327 | 40.9% |
| Men | 80 (10.0%) | 26 (8.0%) | 32.5% |
| Women | 720 (90.0%) | 301 (92.0%) | 41.8% |
| Sources | Recipients | Phylogenetically Strongly Supported Transmission Pairs Including Same-Sex Pairs * (Count, Proportion) | Phylogenetically Strongly Supported Transmission Pairs Excluding Same-Sex Pairs * (Count, Proportion) | Estimated Transmission Flows among Study Participants, Based on Data Including Same-Sex Pairs ** (Mean, 95% Credibility Interval of Posterior Density) | Estimated Transmission Flows among Study Participants, Based on Data Excluding Same-Sex Pairs ** (Mean, 95% Credibility Interval of Posterior Density) | Estimated Transmission Flow Ratios, Based on Data Excluding Same-Sex Pairs *** (Mean, 95% Credibility Interval of Posterior Density) |
|---|---|---|---|---|---|---|
| FF | FF | 33 (31.4%) | 33 (44.6%) | 35.8% (26.2–46.1%) | 45.5% (34.1–57.0%) | -- |
| FF | GP | 9 (8.6%) | 9 (12.1%) | 9.9% (4.7–16.8%) | 12.7% (6.1–21.3%) | -- |
| FF | WHR | 2 (1.9%) | 1 (1.4%) | -- | -- | -- |
| GP | FF | 14 (13.3%) | 14 (18.9%) | 15.3% (8.7–23.3%) | 19.5% (11.4–29.3%) | 1.56 (0.68–3.72) |
| GP | GP | 36 (34.3%) | 16 (21.6%) | 39.0% (29.0–49.4%) | 22.3% (13.4–32.7%) | -- |
| WHR | GP | 1 (1.0%) | 1 (1.4%) | -- | -- | -- |
| WHR | WHR | 10 (9.5%) | 0 (0%) | -- | -- | -- |
| Estimated sources of infection among study participants, based on data excluding same-sex pairs * (mean, 95% credibility interval of posterior density) | ||
| FF | GP | |
| Recipient | ||
| FF | 70.4% (56.2–81.9%) | 29.6 (18.1–43.8%) |
| GP | 19.8% (10.0–33.2%) | 80.2% (66.8–90.0%) |
| Phylogenetically strongly supported transmission pairs excluding same-sex pairs * (count, proportion) | Estimated transmission flows among study participants, based on data excluding same-sex pairs ** (mean, 95% credibility interval of posterior density) | Estimated transmission flow ratios, based on data excluding same-sex pairs *** (mean, 95% credibility interval of posterior density) | ||
| Source | Recipient | |||
| Men 18–24 years | Women 18–24 years | 9 (12.5%) | 9.1% (4.2–15.7%) | 3.16 (0.92–14.44) |
| Men 18–24 years | Women 25–59 years | 9 (12.5%) | 11.7% (5.6–19.9%) | 1.29 (0.47–3.69) |
| Men 25–59 years | Women 18–24 years | 13 (18.1 %) | 17.0% (9.2–26.3%) | 2.63 (0.96–8.28) |
| Men 25–59 years | Women 25–59 years | 18 (25%) | 29.8% (19.2–41.7%) | 2.29 (1.02–5.52) |
| Women 18–24 years | Men 18–24 years | 3 (4.2%) | 3.1% (0.7–7.5%) | -- |
| Women 18–24 years | Men 25–59 years | 5 (6.9%) | 6.7% (2.3–13.4%) | -- |
| Women 25–59 years | Men 18–24 years | 7 (9.7%) | 9.2% (3.8–16.8%) | -- |
| Women 25–59 years | Men 25–59 years | 8 (11.1%) | 13.4% (6.1–22.9%) | -- |
| Estimated sources of infection among study participants, based on data excluding same-sex pairs * (mean, 95% credibility interval of posterior density) | ||||
| Men 18–24 years | Men 25–59 years | Women 18–24 years | Women 25–59 years | |
| Recipient | ||||
| Women 18–24 years | 34.7% (17.1–55.9% | 65.3% (44.1–82.9%) | -- | -- |
| Women 25–59 years | 27.7% (14.1–45.7%) | 72.3% (54.3–85.9%) | -- | -- |
| Men 18–24 years | -- | -- | 24.2% (6.3–54.0%) | 75.8% (46.0–93.7%) |
| Men 25–59 years | -- | -- | 32.6% (12.7–59.9%) | 67.4% (40.1–87.3%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bbosa, N.; Ssemwanga, D.; Ssekagiri, A.; Xi, X.; Mayanja, Y.; Bahemuka, U.; Seeley, J.; Pillay, D.; Abeler-Dörner, L.; Golubchik, T.; et al. Phylogenetic and Demographic Characterization of Directed HIV-1 Transmission Using Deep Sequences from High-Risk and General Population Cohorts/Groups in Uganda. Viruses 2020, 12, 331. https://doi.org/10.3390/v12030331
Bbosa N, Ssemwanga D, Ssekagiri A, Xi X, Mayanja Y, Bahemuka U, Seeley J, Pillay D, Abeler-Dörner L, Golubchik T, et al. Phylogenetic and Demographic Characterization of Directed HIV-1 Transmission Using Deep Sequences from High-Risk and General Population Cohorts/Groups in Uganda. Viruses. 2020; 12(3):331. https://doi.org/10.3390/v12030331
Chicago/Turabian StyleBbosa, Nicholas, Deogratius Ssemwanga, Alfred Ssekagiri, Xiaoyue Xi, Yunia Mayanja, Ubaldo Bahemuka, Janet Seeley, Deenan Pillay, Lucie Abeler-Dörner, Tanya Golubchik, and et al. 2020. "Phylogenetic and Demographic Characterization of Directed HIV-1 Transmission Using Deep Sequences from High-Risk and General Population Cohorts/Groups in Uganda" Viruses 12, no. 3: 331. https://doi.org/10.3390/v12030331
APA StyleBbosa, N., Ssemwanga, D., Ssekagiri, A., Xi, X., Mayanja, Y., Bahemuka, U., Seeley, J., Pillay, D., Abeler-Dörner, L., Golubchik, T., Fraser, C., Kaleebu, P., Ratmann, O., & on behalf of the MRC/UVRI & LSHTM Uganda Research Unit and The PANGEA Consortium. (2020). Phylogenetic and Demographic Characterization of Directed HIV-1 Transmission Using Deep Sequences from High-Risk and General Population Cohorts/Groups in Uganda. Viruses, 12(3), 331. https://doi.org/10.3390/v12030331