In Search of Covariates of HIV-1 Subtype B Spread in the United States—A Cautionary Tale of Large-Scale Bayesian Phylogeography

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Data
2.2. Subtyping
2.3. Multiple Sequence Alignment
2.4. Subsampling Strategy
2.5. Phylogenetic Inference
2.6. Geographic History Reconstruction
3. Results
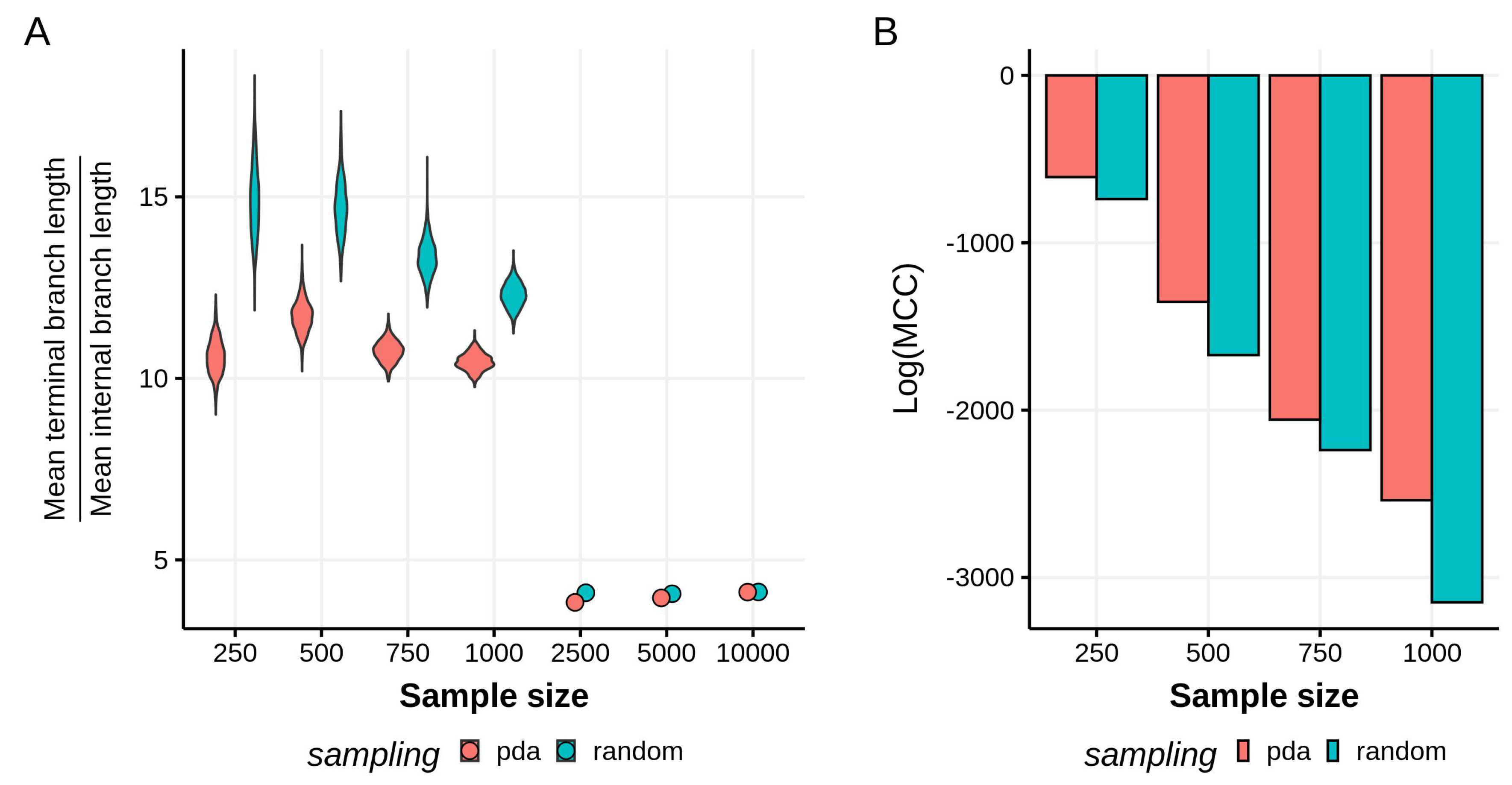
3.1. Effects of Sampling Scheme on Reconstructed Tree Topologies
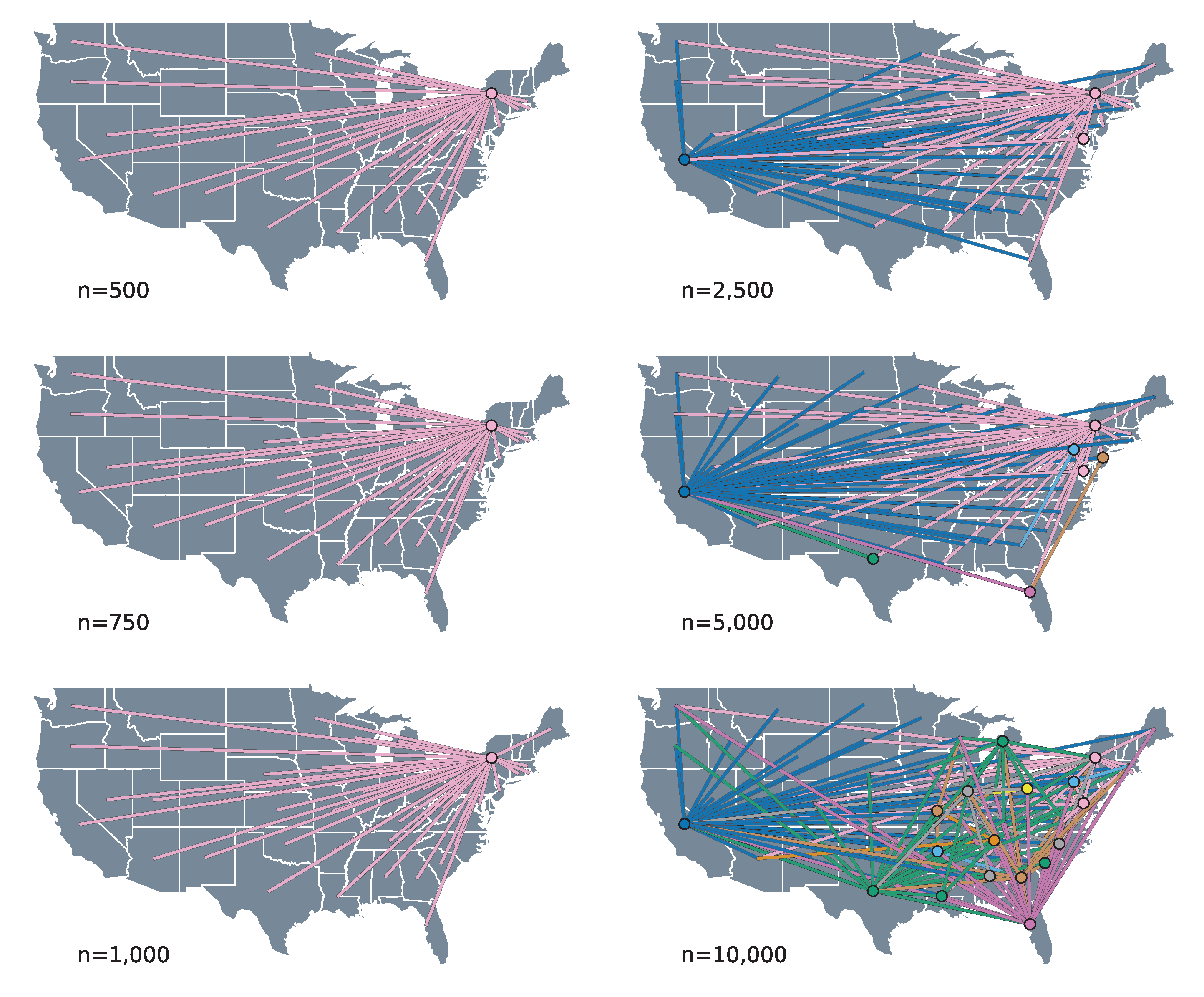
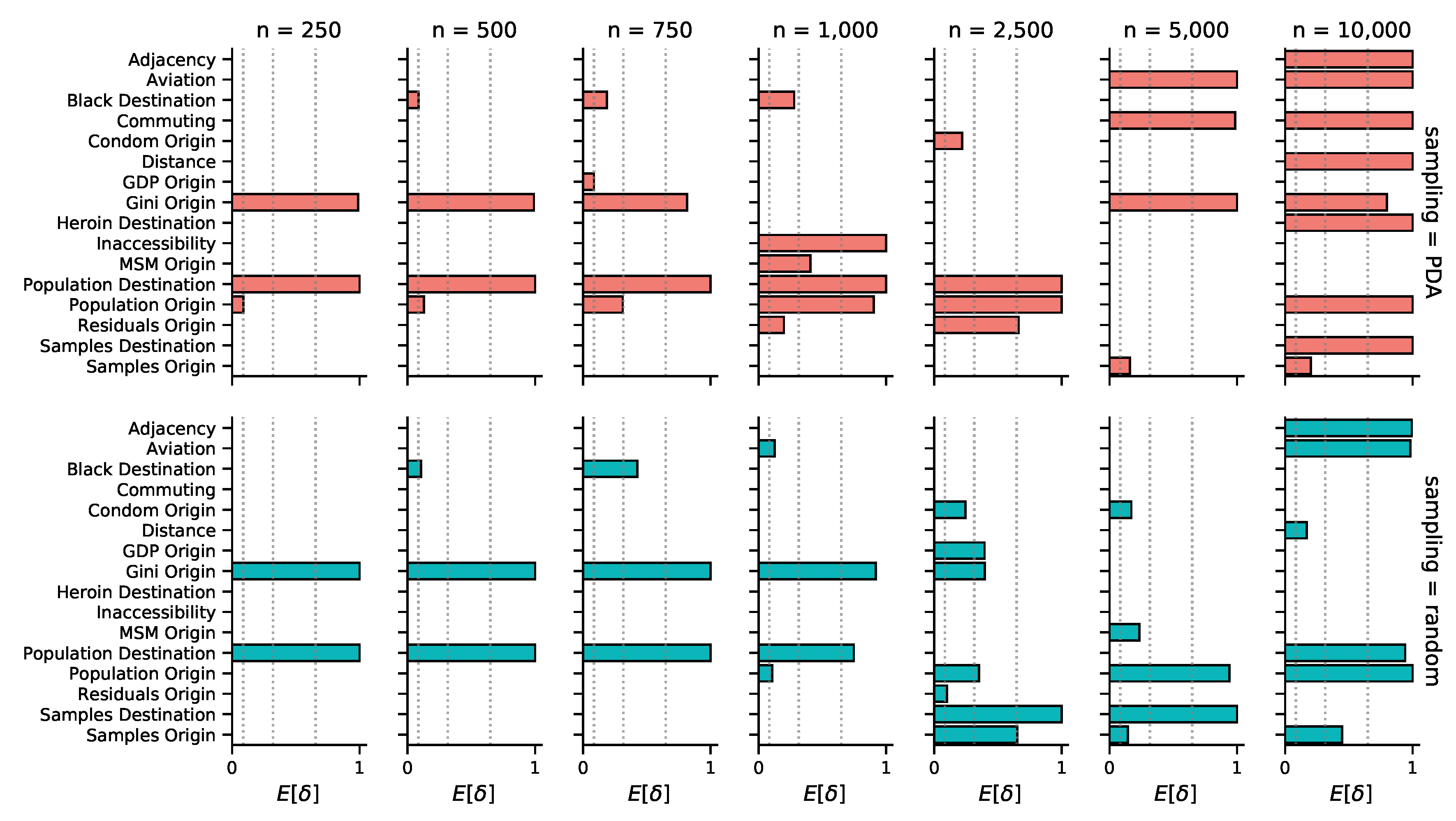
3.2. Phylogeographic Reconstruction and Drivers of Spread
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The next-generation sequencing revolution and its impact on genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Kao, R.R.; Haydon, D.T.; Lycett, S.J.; Murcia, P.R. Supersize me: How whole-genome sequencing and big data are transforming epidemiology. Trends Microbiol. 2014, 22, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.M.; Romero-Severson, E.; Leitner, T. Phylodynamic Inference across Epidemic Scales. Mol. Biol. Evol. 2017, 34, 1276–1288. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vrancken, B.; Rambaut, A.; Suchard, M.A.; Drummond, A.; Baele, G.; Derdelinckx, I.; Van Wijngaerden, E.; Vandamme, A.M.; Van Laethem, K.; Lemey, P. The genealogical population dynamics of HIV-1 in a large transmission chain: Bridging within and among host evolutionary rates. PLoS Comput. Biol. 2014, 10, e1003505. [Google Scholar] [CrossRef]
- Grenfell, B.T.; Pybus, O.G.; Gog, J.R.; Wood, J.L.N.; Daly, J.M.; Mumford, J.A.; Holmes, E.C. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 2004, 303, 327–332. [Google Scholar] [CrossRef]
- Holmes, E.C. The phylogeography of human viruses. Mol. Ecol. 2004, 13, 745–756. [Google Scholar] [CrossRef]
- Faria, N.R.; Suchard, M.A.; Rambaut, A.; Lemey, P. Toward a quantitative understanding of viral phylogeography. Curr. Opin. Virol. 2011, 1, 423–429. [Google Scholar] [CrossRef]
- Biek, R.; Pybus, O.G.; Lloyd-Smith, J.O.; Didelot, X. Measurably evolving pathogens in the genomic era. Trends Ecol. Evol. 2015, 30, 306–313. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Welch, J.J.; Suchard, M.A. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 2010, 27, 1877–1885. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Bedford, T.; Faria, N.; Bielejec, F.; Baele, G.; Russell, C.A.; Smith, D.J.; Pybus, O.G.; Brockmann, D.; et al. Unifying viral genetics and human transportation data to predict the global transmission dynamics of human influenza H3N2. PLoS Pathog. 2014, 10, e1003932. [Google Scholar] [CrossRef] [PubMed]
- Dellicour, S.; Rose, R.; Pybus, O.G. Explaining the geographic spread of emerging epidemics: A framework for comparing viral phylogenies and environmental landscape data. BMC Bioinform. 2016, 17, 82. [Google Scholar] [CrossRef] [PubMed]
- Vrancken, B.; Adachi, D.; Benedet, M.; Singh, A.; Read, R.; Shafran, S.; Taylor, G.D.; Simmonds, K.; Sikora, C.; Lemey, P.; et al. The multi-faceted dynamics of HIV-1 transmission in Northern Alberta: A combined analysis of virus genetic and public health data. Infect. Genet. Evol. 2017, 52, 100–105. [Google Scholar] [CrossRef]
- Al-Qahtani, A.A.; Baele, G.; Khalaf, N.; Suchard, M.A.; Al-Anazi, M.R.; Abdo, A.A.; Sanai, F.M.; Al-Ashgar, H.I.; Khan, M.Q.; Al-Ahdal, M.N.; et al. The epidemic dynamics of hepatitis C virus subtypes 4a and 4d in Saudi Arabia. Sci Rep 2017, 7, 44947. [Google Scholar] [CrossRef]
- Cuypers, L.; Vrancken, B.; Fabeni, L.; Marascio, N.; Cento, V.; Di Maio, V.C.; Aragri, M.; Pineda-Peña, A.C.; Schrooten, Y.; Van Laethem, K.; et al. Implications of hepatitis C virus subtype 1a migration patterns for virus genetic sequencing policies in Italy. BMC Evol. Biol. 2017, 17, 70. [Google Scholar] [CrossRef]
- Pérez, A.B.; Vrancken, B.; Chueca, N.; Aguilera, A.; Reina, G.; García-Del Toro, M.; Vera, F.; Von Wichman, M.A.; Arenas, J.I.; Téllez, F.; et al. Increasing importance of European lineages in seeding the hepatitis C virus subtype 1a epidemic in Spain. Eurosurveillance 2019, 24. [Google Scholar] [CrossRef]
- Vrancken, B.; Cuypers, L.; Pérez, A.B.; Chueca, N.; Anton-Basantas, J.; de la Iglesia, A.; Fuentes, J.; Pineda, J.A.; Téllez, F.; Bernal, E.; et al. Cross-country migration linked to people who inject drugs challenges the long-term impact of national HCV elimination programmes. J. Hepatol. 2019, 71, 1270–1272. [Google Scholar] [CrossRef]
- Talbi, C.; Lemey, P.; Suchard, M.A.; Abdelatif, E.; Elharrak, M.; Nourlil, J.; Faouzi, A.; Echevarría, J.E.; Vazquez Morón, S.; Rambaut, A.; et al. Phylodynamics and human-mediated dispersal of a zoonotic virus. PLoS Pathog. 2010, 6, e1001166. [Google Scholar] [CrossRef]
- Nelson, M.I.; Lemey, P.; Tan, Y.; Vincent, A.; Lam, T.T.Y.; Detmer, S.; Viboud, C.; Suchard, M.A.; Rambaut, A.; Holmes, E.C.; et al. Spatial dynamics of human-origin H1 influenza A virus in North American swine. PLoS Pathog. 2011, 7, e1002077. [Google Scholar] [CrossRef]
- Faria, N.R.; Rambaut, A.; Suchard, M.A.; Baele, G.; Bedford, T.; Ward, M.J.; Tatem, A.J.; Sousa, J.D.; Arinaminpathy, N.; Pépin, J.; et al. HIV epidemiology. The early spread and epidemic ignition of HIV-1 in human populations. Science 2014, 346, 56–61. [Google Scholar] [CrossRef]
- Magiorkinis, G.; Angelis, K.; Mamais, I.; Katzourakis, A.; Hatzakis, A.; Albert, J.; Lawyer, G.; Hamouda, O.; Struck, D.; Vercauteren, J.; et al. The global spread of HIV-1 subtype B epidemic. Infect. Genet. Evol. 2016, 46, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Watts, T.D.; McKay, R.A.; Suchard, M.A.; Granade, T.; Teuwen, D.E.; Koblin, B.A.; Heneine, W.; Lemey, P.; Jaffe, H.W. 1970s and ’Patient 0’ HIV-1 genomes illuminate early HIV/AIDS history in North America. Nature 2016, 539, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Rambaut, A. Many-core algorithms for statistical phylogenetics. Bioinformatics 2009, 25, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Ayres, D.L.; Darling, A.; Zwickl, D.J.; Beerli, P.; Holder, M.T.; Lewis, P.O.; Huelsenbeck, J.P.; Ronquist, F.; Swofford, D.L.; Cummings, M.P.; et al. BEAGLE: An application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol. 2012, 61, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Lemey, P.; Rambaut, A.; Suchard, M.A. Adaptive MCMC in Bayesian phylogenetics: An application to analyzing partitioned data in BEAST. Bioinformatics 2017, 33, 1798–1805. [Google Scholar] [CrossRef] [PubMed]
- Ayres, D.L.; Cummings, M.P.; Baele, G.; Darling, A.E.; Lewis, P.O.; Swofford, D.L.; Huelsenbeck, J.P.; Lemey, P.; Rambaut, A.; Suchard, M.A. BEAGLE 3: Improved Performance, Scaling, and Usability for a High-Performance Computing Library for Statistical Phylogenetics. Syst. Biol. 2019, 68, 1052–1061. [Google Scholar] [CrossRef]
- Pyne, M.T.; Hackett, J., Jr.; Holzmayer, V.; Hillyard, D.R. Large-scale analysis of the prevalence and geographic distribution of HIV-1 non-B variants in the United States. J. Clin. Microbiol. 2013, 51, 2662–2669. [Google Scholar] [CrossRef]
- Pineda-Peña, A.C.; Faria, N.R.; Imbrechts, S.; Libin, P.; Abecasis, A.B.; Deforche, K.; Gómez-López, A.; Camacho, R.J.; de Oliveira, T.; Vandamme, A.M. Automated subtyping of HIV-1 genetic sequences for clinical and surveillance purposes: Performance evaluation of the new REGA version 3 and seven other tools. Infect. Genet. Evol. 2013, 19, 337–348. [Google Scholar] [CrossRef]
- Libin, P.J.K.; Deforche, K.; Abecasis, A.B.; Theys, K. VIRULIGN: Fast codon-correct alignment and annotation of viral genomes. Bioinformatics 2019, 35, 1763–1765. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 2010, 5, e9490. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
- HIV Surveillance Reports Archive | Reports| Resource Library | HIV/AIDS | CDC. 2019. Available online: https://www.cdc.gov/hiv/library/reports/hiv-surveillance-archive.html (accessed on 21 July 2019).
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Chernomor, O.; Minh, B.Q.; Forest, F.; Klaere, S.; Ingram, T.; Henzinger, M.; von Haeseler, A. Split diversity in constrained conservation prioritization using integer linear programming. Methods Ecol. Evol. 2015, 6, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Yang, Z. Maximum-likelihood estimation of phylogeny from DNA sequences when substitution rates differ over sites. Mol. Biol. Evol. 1993, 10, 1396–1401. [Google Scholar]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Dudas, G.; Carvalho, L.M.; Bedford, T.; Tatem, A.J.; Baele, G.; Faria, N.R.; Park, D.J.; Ladner, J.T.; Arias, A.; Asogun, D.; et al. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 2017, 544, 309–315. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Pybus, O.G. HIV evolutionary dynamics within and among hosts. AIDS Rev. 2006, 8, 125–140. [Google Scholar] [PubMed]
- Volz, E.M.; Koelle, K.; Bedford, T. Viral phylodynamics. PLoS Comput. Biol. 2013, 9, e1002947. [Google Scholar] [CrossRef] [PubMed]
- Colijn, C.; Plazzotta, G. A Metric on Phylogenetic Tree Shapes. Syst. Biol. 2018, 67, 113–126. [Google Scholar] [CrossRef]
- Duchêne, S.; Duchêne, D.A.; Geoghegan, J.L.; Dyson, Z.A.; Hawkey, J.; Holt, K.E. Inferring demographic parameters in bacterial genomic data using Bayesian and hybrid phylogenetic methods. BMC Evol. Biol. 2018, 18, 95. [Google Scholar] [CrossRef]
- Robbins, K.E.; Lemey, P.; Pybus, O.G.; Jaffe, H.W.; Youngpairoj, A.S.; Brown, T.M.; Salemi, M.; Vandamme, A.M.; Kalish, M.L. U.S. Human immunodeficiency virus type 1 epidemic: Date of origin, population history, and characterization of early strains. J. Virol. 2003, 77, 6359–6366. [Google Scholar] [CrossRef][Green Version]
- Kosakovsky Pond, S.L.; Weaver, S.; Leigh Brown, A.J.; Wertheim, J.O. HIV-TRACE (TRAnsmission Cluster Engine): A Tool for Large Scale Molecular Epidemiology of HIV-1 and Other Rapidly Evolving Pathogens. Mol. Biol. Evol. 2018, 35, 1812–1819. [Google Scholar] [CrossRef]
- HIV Molecular Surveillance: Questions and Answers | Guidance | Program Resources | HIV/AIDS | CDC. 2018. Available online: https://www.cdc.gov/hiv/programresources/guidance/molecular-cluster-identification/qa.html (accessed on 22 July 2019).




{kind=link}
{kind=link}
{kind=link}
| Predictor | Times Included (BF>5) | Bayes Factor | Coefficient |
|---|---|---|---|
| Population size at origin | 10/14 | 265.4 (5.2 - ∞) | 2.8 (1.4 - 3.5) |
| Population size at destination | 10/14 | ∞ (160.1 - ∞) | 1.0 (0.8 - 1.6) |
| Gini index at origin | 10/14 | 5165.4 (36.0 - ∞) | 2.7 (1.0 - 4.2) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, S.L.; Dellicour, S.; Vrancken, B.; Suchard, M.A.; Pyne, M.T.; Hillyard, D.R.; Lemey, P.; Baele, G. In Search of Covariates of HIV-1 Subtype B Spread in the United States—A Cautionary Tale of Large-Scale Bayesian Phylogeography. Viruses 2020, 12, 182. https://doi.org/10.3390/v12020182
Hong SL, Dellicour S, Vrancken B, Suchard MA, Pyne MT, Hillyard DR, Lemey P, Baele G. In Search of Covariates of HIV-1 Subtype B Spread in the United States—A Cautionary Tale of Large-Scale Bayesian Phylogeography. Viruses. 2020; 12(2):182. https://doi.org/10.3390/v12020182
Chicago/Turabian StyleHong, Samuel L., Simon Dellicour, Bram Vrancken, Marc A. Suchard, Michael T. Pyne, David R. Hillyard, Philippe Lemey, and Guy Baele. 2020. "In Search of Covariates of HIV-1 Subtype B Spread in the United States—A Cautionary Tale of Large-Scale Bayesian Phylogeography" Viruses 12, no. 2: 182. https://doi.org/10.3390/v12020182
APA StyleHong, S. L., Dellicour, S., Vrancken, B., Suchard, M. A., Pyne, M. T., Hillyard, D. R., Lemey, P., & Baele, G. (2020). In Search of Covariates of HIV-1 Subtype B Spread in the United States—A Cautionary Tale of Large-Scale Bayesian Phylogeography. Viruses, 12(2), 182. https://doi.org/10.3390/v12020182