Effects of TDP2/VPg Unlinkase Activity on Picornavirus Infections Downstream of Virus Translation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus Stocks
2.2. Generation of 35S-Methionine-Labeled VPg-Linked RNA Substrate and VPg Unlinkase Assay
2.3. Generation of Tagged TDP2-Expressing hRPE Cell Lines
2.4. Virus Infections and Single Cycle Growth Analysis
2.5. Preparation of Lysates from Uninfected and Infected Cells and Western Blot Analysis
2.6. Generation of Renilla Luciferase Reporter Virus and Luciferase Assays
2.7. Polysome Profile Analysis and Quantitation of Viral RNA Production
2.8. Encapsidation Inhibition Assays
2.9. Immunofluorescence Assays
3. Results
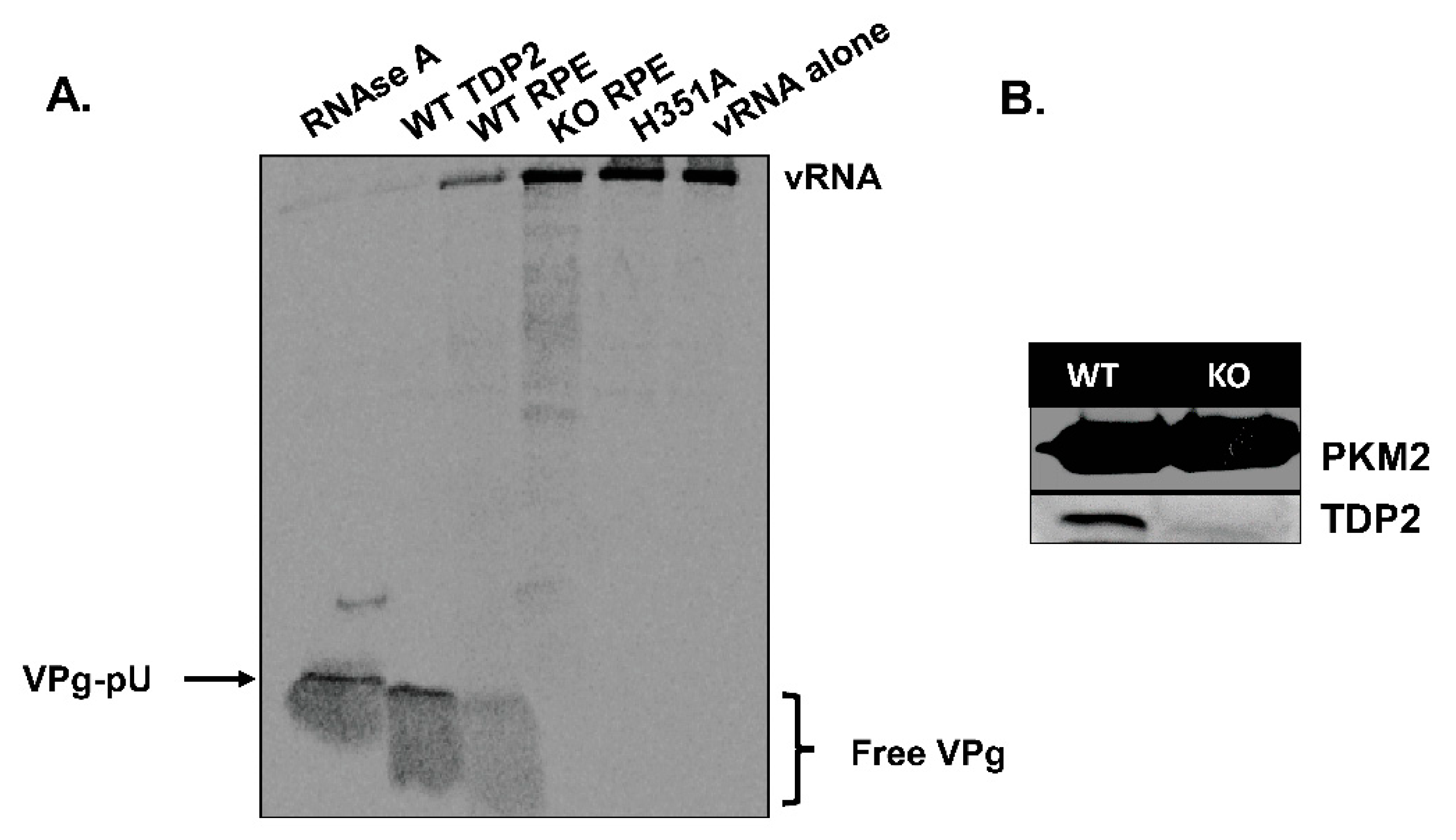
3.1. TDP2 Protein Expression and VPg Unlinkase Activity Are Absent in TDP2 Knock-Out hRPE-1 Cells
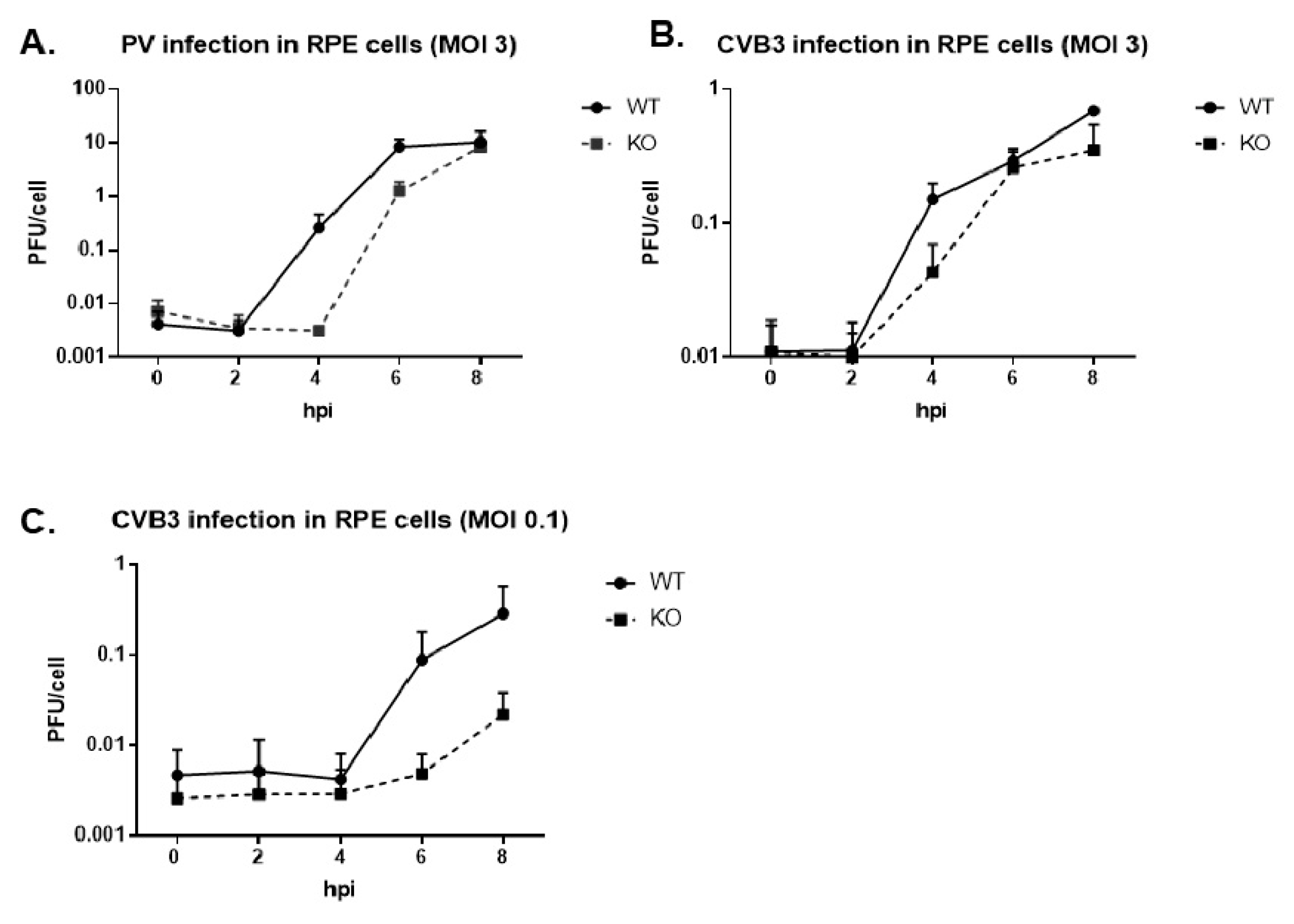
3.2. Virus Growth Kinetics Are Delayed in the Absence of TDP2 for Poliovirus and CVB3 in hRPE-1 Cells, and the Effect is MOI-Dependent
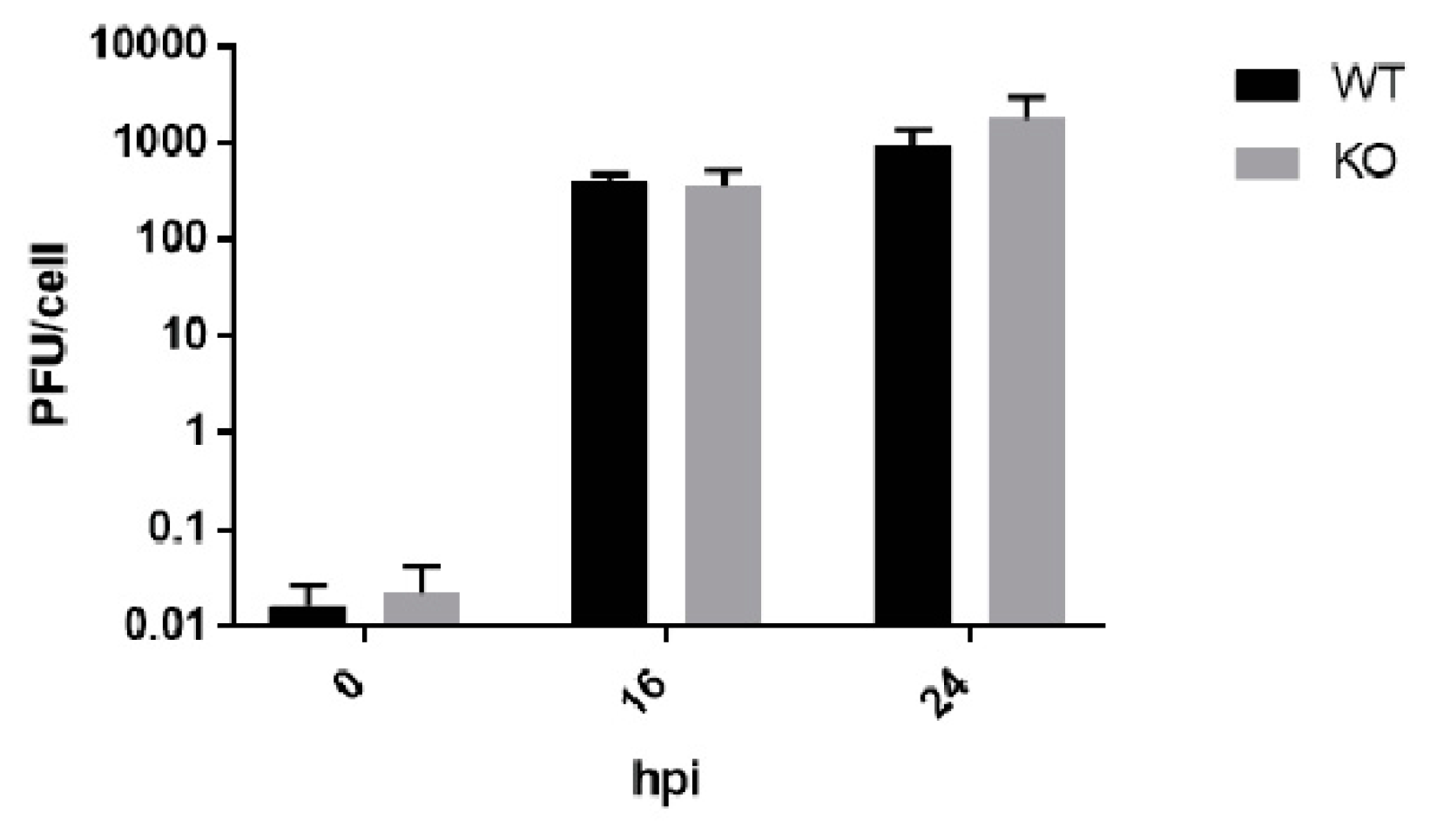
3.3. Poliovirus Titers Are Comparable in WT and KO RPE cells after Multiple Replication Cycles
3.4. eIF4G is Cleaved in both WT and KO TDP2 RPE Cells during Poliovirus Infection
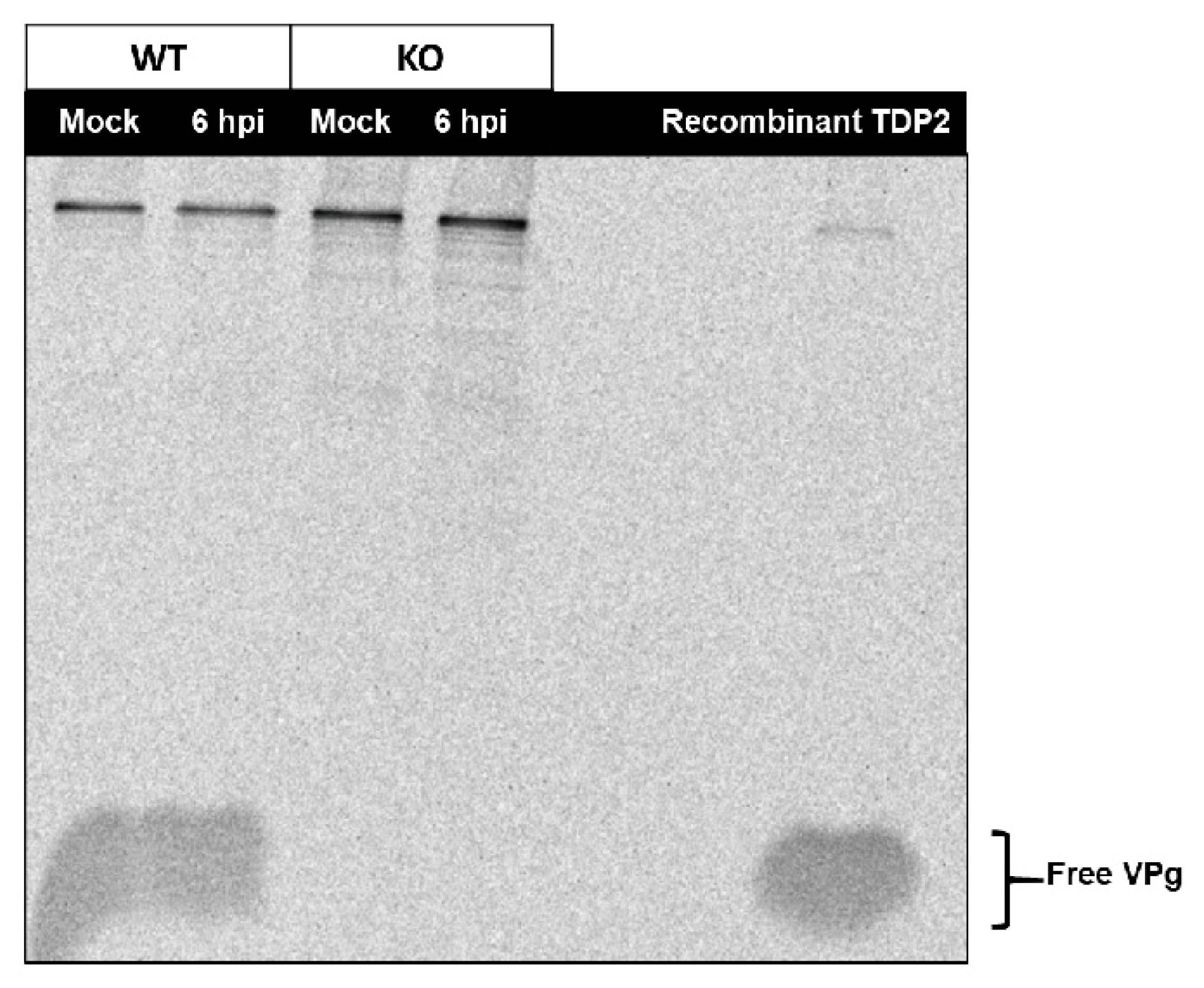
3.5. An Alternative Source of VPg Unlinkase Activity Is Not Activated at Late Times of Infection in TDP2 KO hRPE-1 Cells
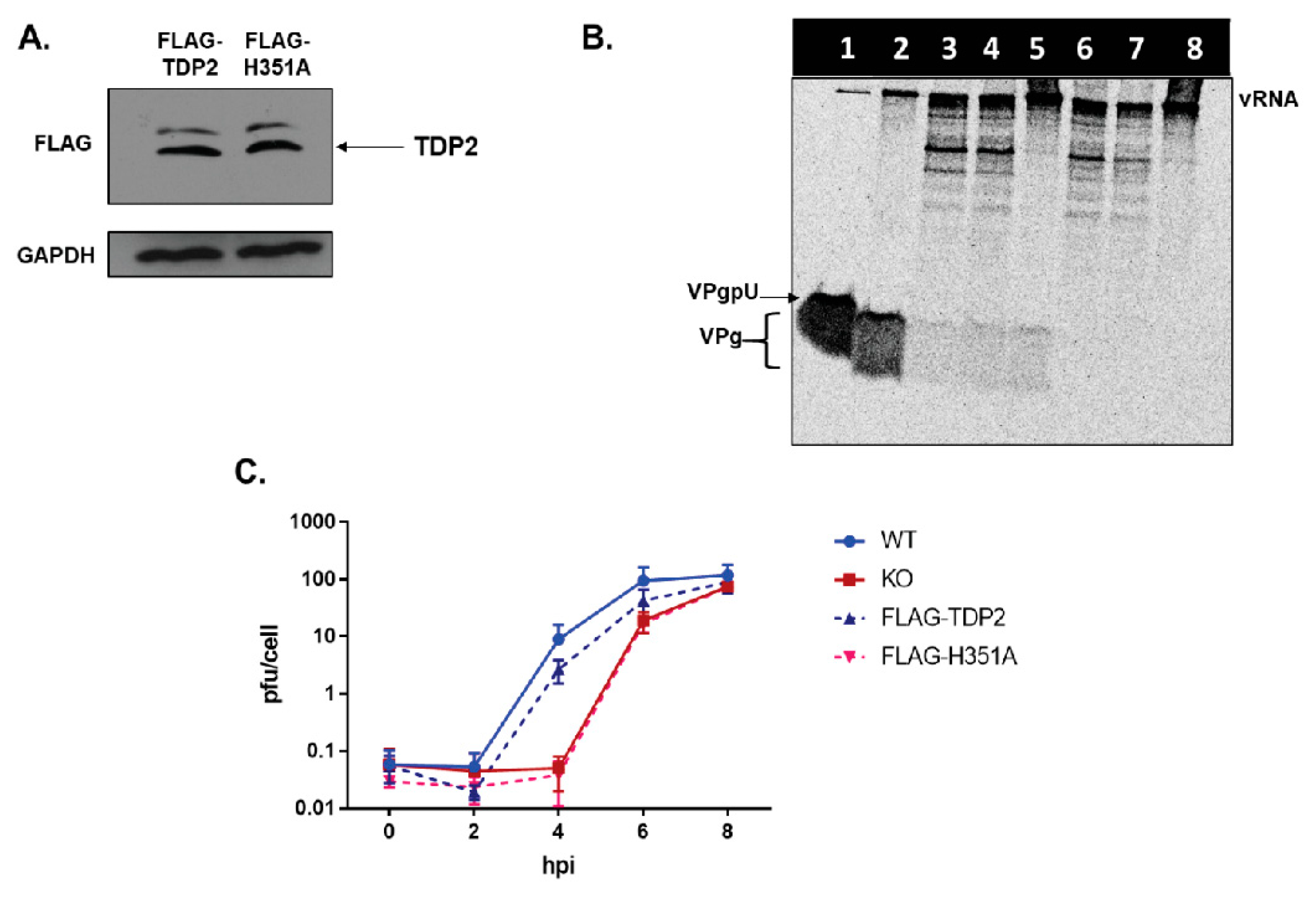
3.6. TDP2 5′ Phosphodiesterase Activity Is Required for Efficient Poliovirus Replication
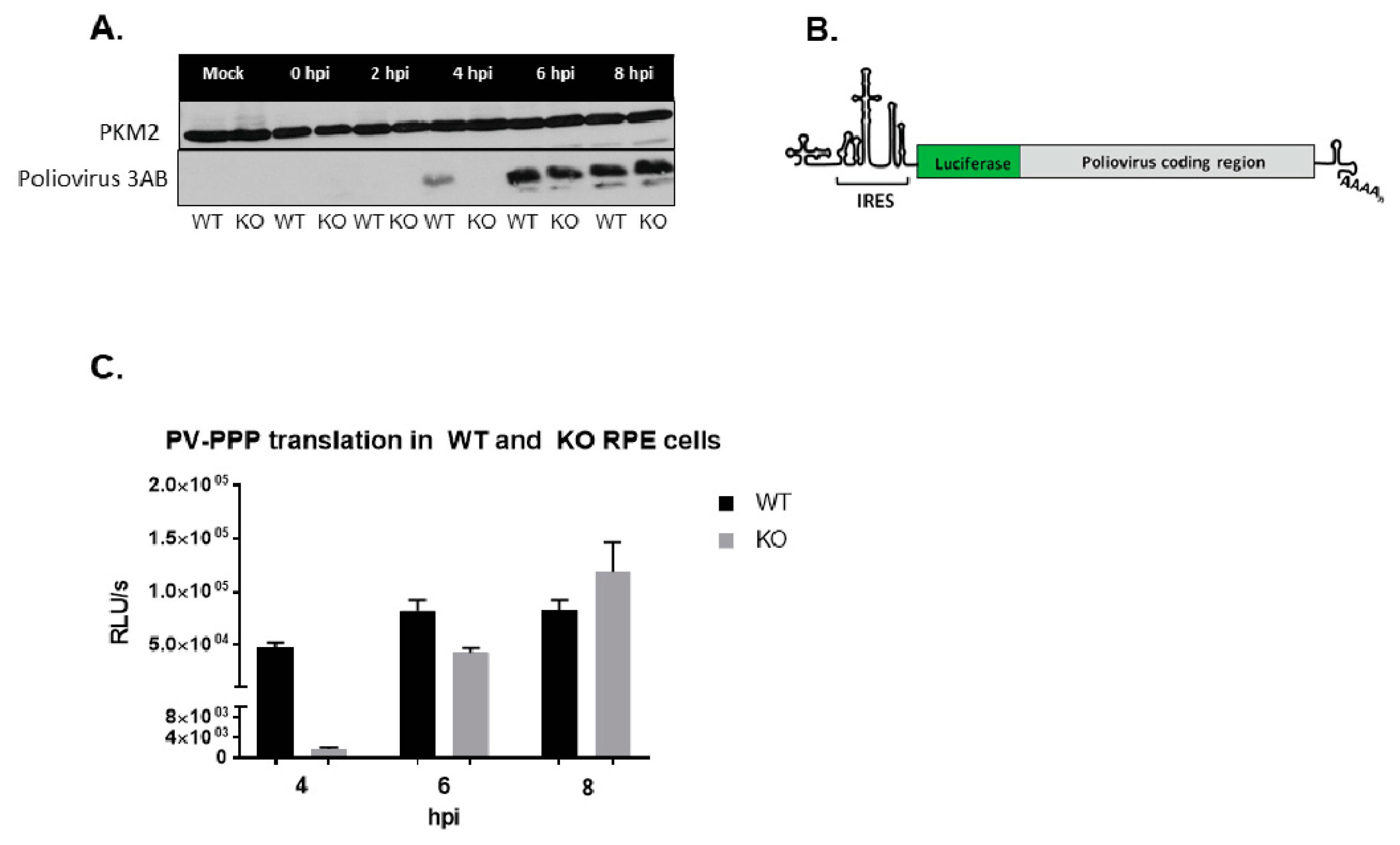
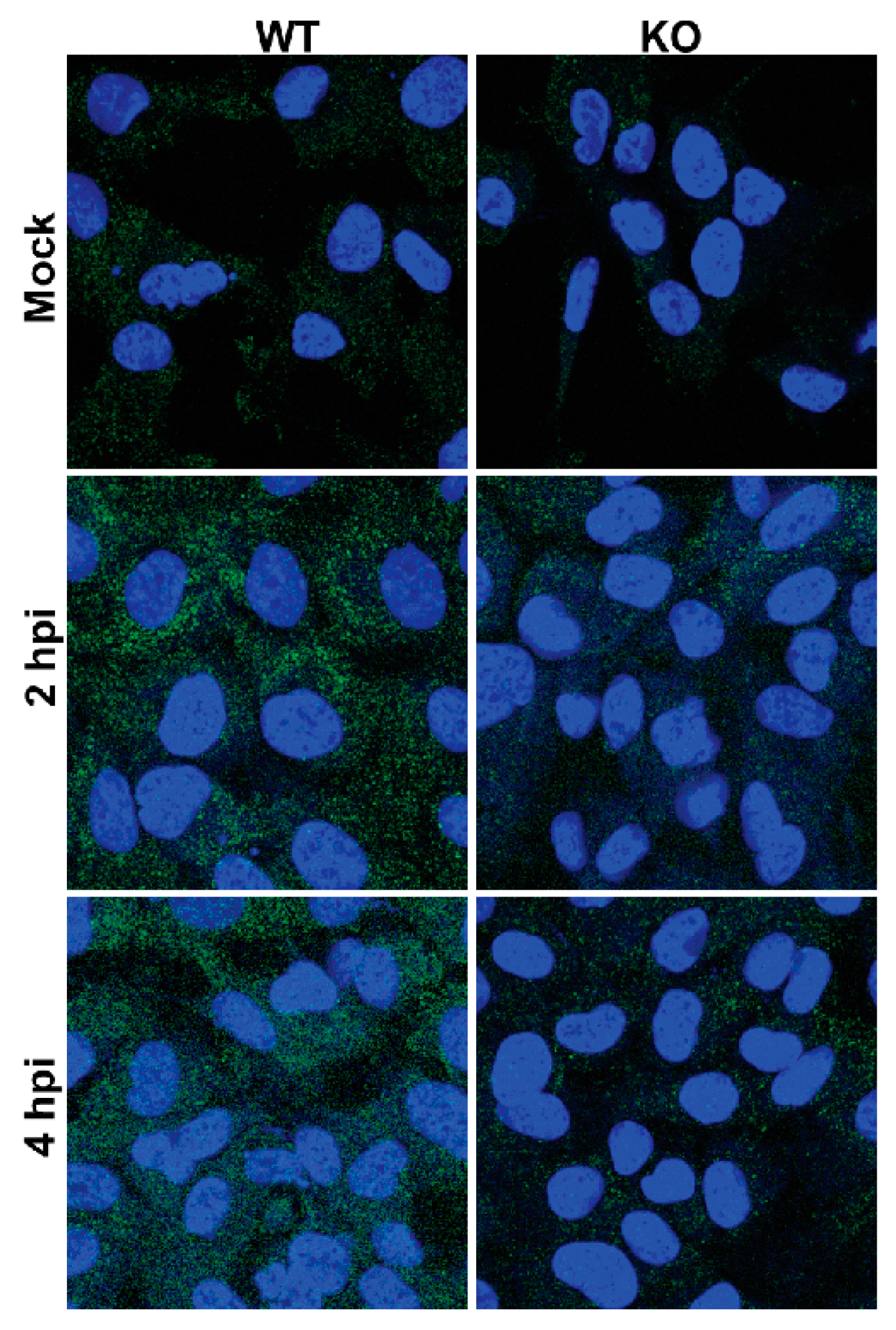
3.7. Poliovirus Protein Production Is Delayed in the Absence of TDP2 in hRPE-1 Cells
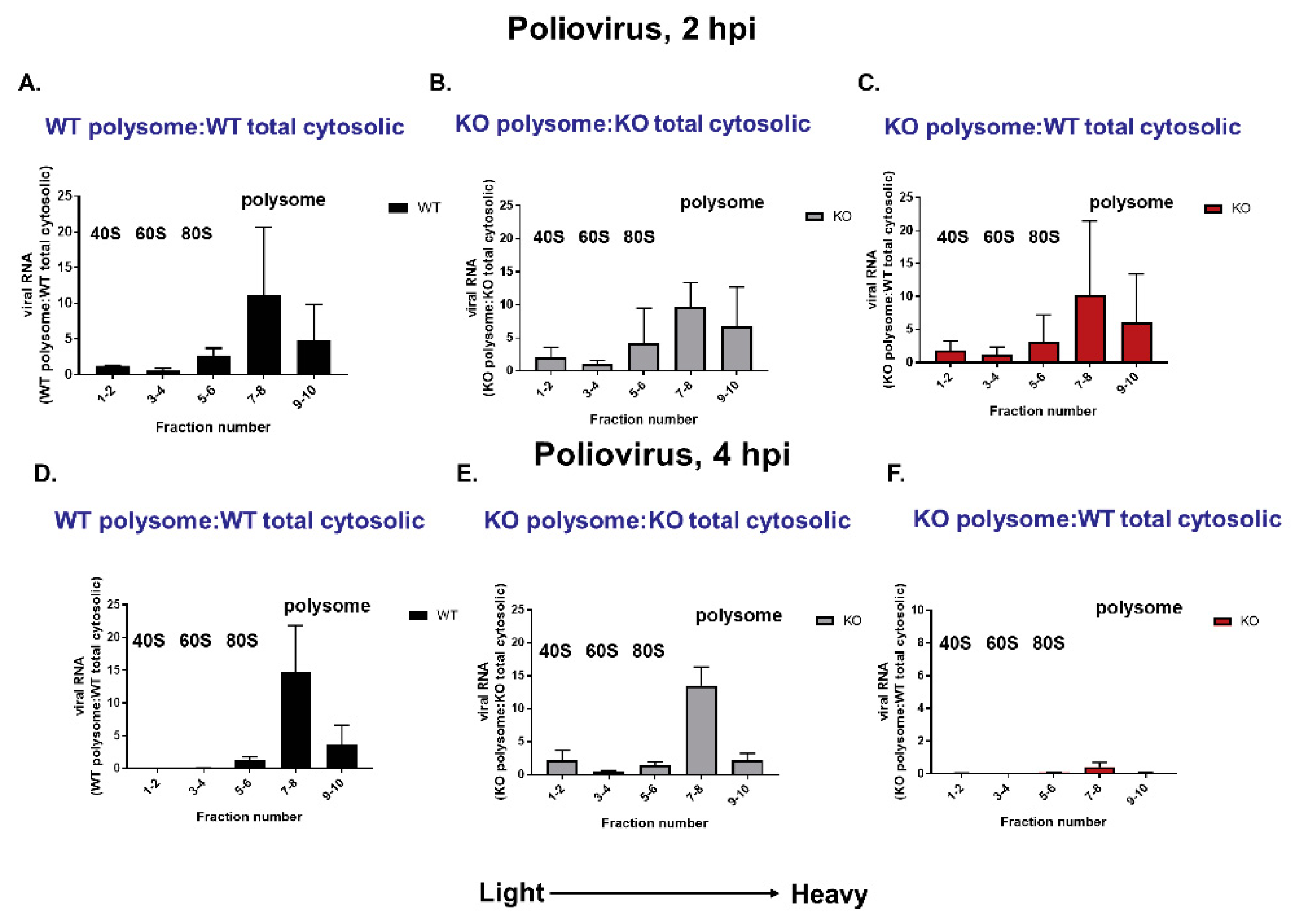
3.8. Polysomes Can Assemble Efficiently on VPg-Linked RNA after the Initial Round of Translation and Replication in Cultured Cells
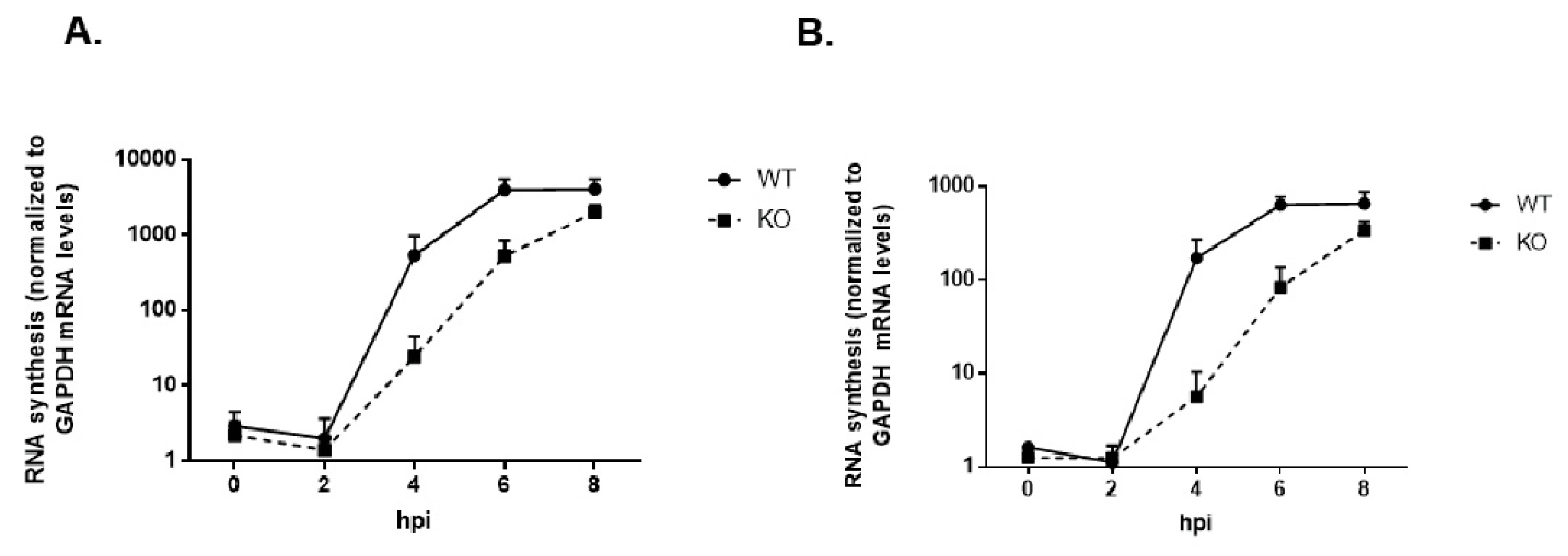
3.9. Viral RNA Synthesis Is Delayed in the Absence of TDP2 in hRPE Cells
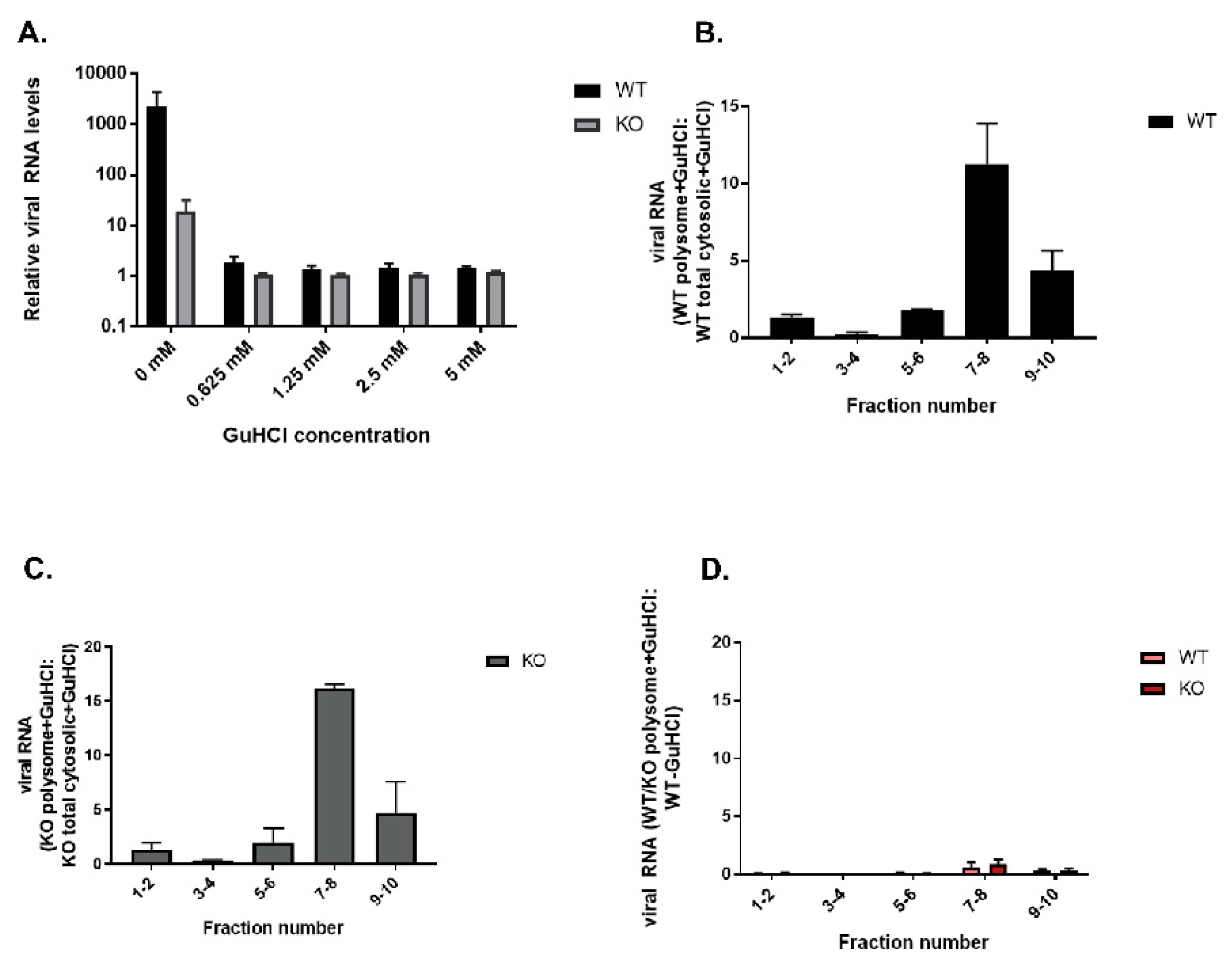
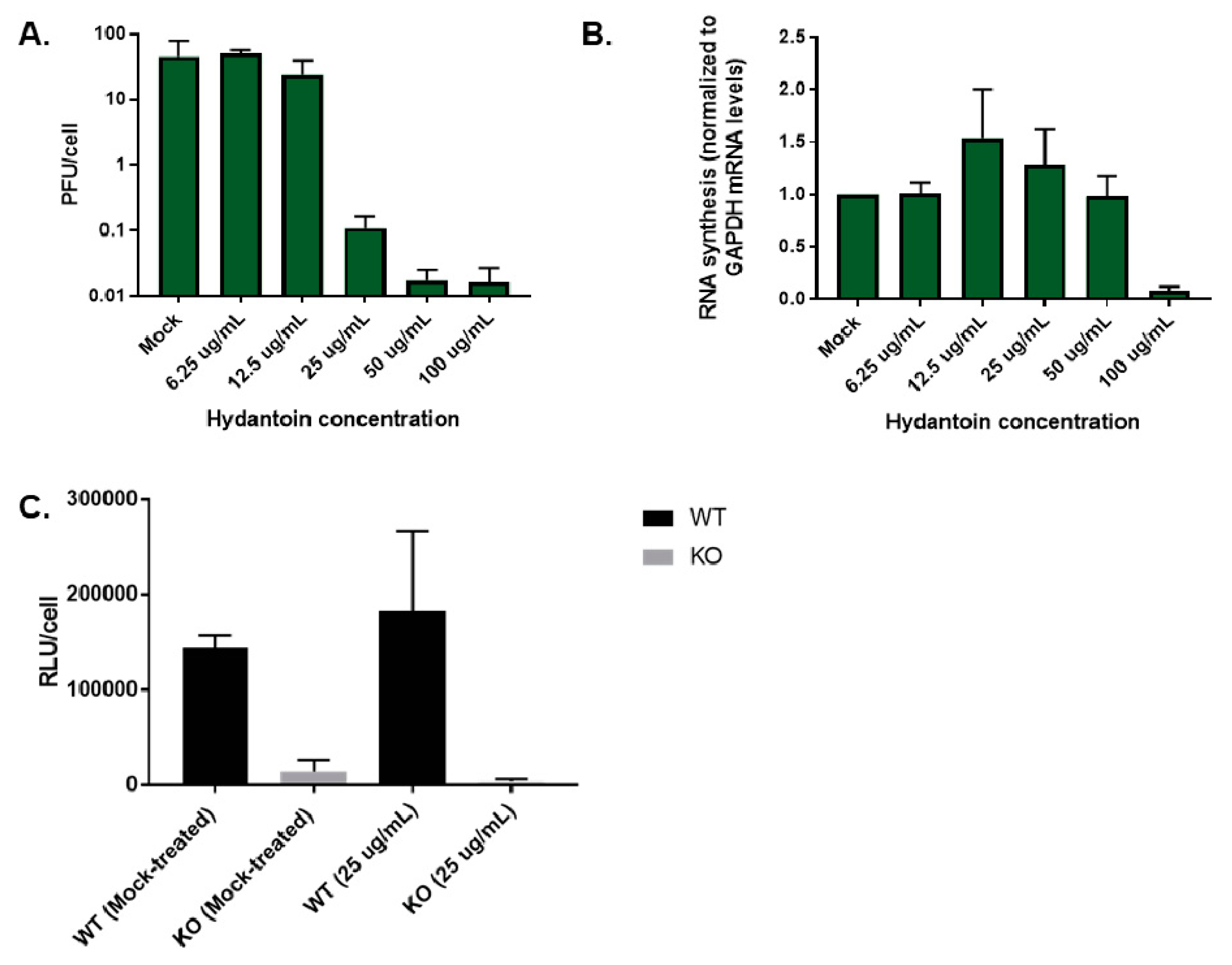
3.10. Premature Encapsidation of Viral RNA Is not Responsible for Observed Growth Phenotype in the Absence of TDP2
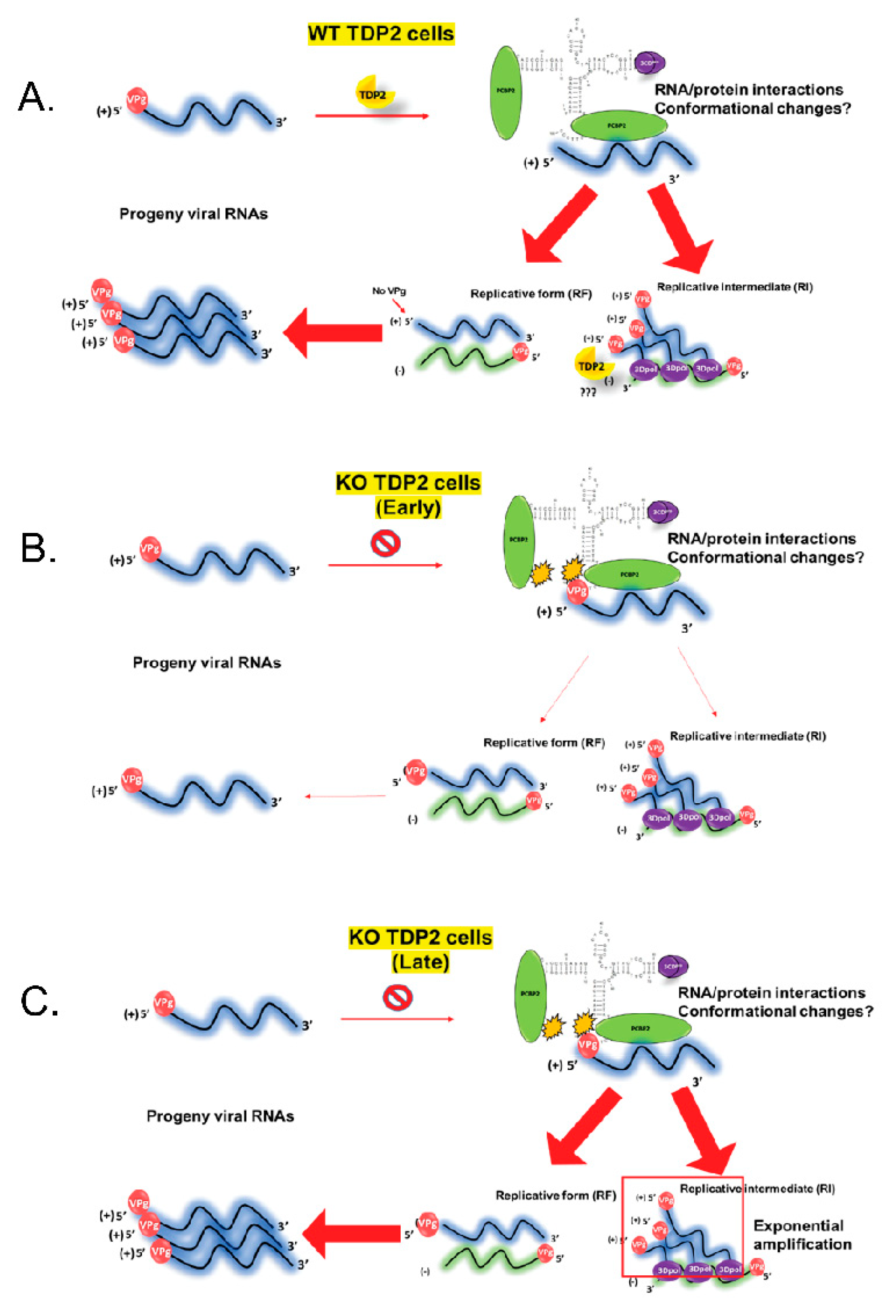
3.11. Replicative Form (RF) and/or Replicative Intermediate (RI) Formation Is Delayed in TDP2 KO hRPE-1 Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singanayagam, A.; Joshi, P.V.; Mallia, P.; Johnston, S.L. Viruses exacerbating chronic pulmonary disease: The role of immune modulation. BMC Med. 2012, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Messacar, K.; Abzug, M.J.; Dominguez, S.R. The Emergence of Enterovirus-D68. Microbiol Spectr 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Dyda, A.; Stelzer-Braid, S.; Adam, D.; Chughtai, A.A.; MacIntyre, C.R. The association between acute flaccid myelitis (AFM) and Enterovirus D68 (EV-D68)—what is the evidence for causation? Euro Surveill 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y. Enterovirus 71 infection and neurological complications. Korean J. Pediatr. 2016, 59, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.V.; van Boom, J.H.; Filippov, D.; Wimmer, E. Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nature 1998, 393, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Gerber, K.; Wimmer, E.; Paul, A.V. Biochemical and genetic studies of the initiation of human rhinovirus 2 RNA replication: Identification of a cis-replicating element in the coding sequence of 2A(pro). J. Virol. 2001, 75, 10979–10990. [Google Scholar] [CrossRef]
- Goodfellow, I.; Chaudhry, Y.; Richardson, A.; Meredith, J.; Almond, J.W.; Barclay, W.; Evans, D.J. Identification of a cis-acting replication element within the poliovirus coding region. J. Virol. 2000, 74, 4590–4600. [Google Scholar] [CrossRef]
- McKnight, K.L.; Lemon, S.M. The rhinovirus type 14 genome contains an internally located RNA structure that is required for viral replication. RNA 1998, 4, 1569–1584. [Google Scholar] [CrossRef]
- Goodfellow, I.G.; Polacek, C.; Andino, R.; Evans, D.J. The poliovirus 2C cis-acting replication element-mediated uridylylation of VPg is not required for synthesis of negative-sense genomes. J. Gen. Virol. 2003, 84, 2359–2363. [Google Scholar] [CrossRef]
- Steil, B.P.; Barton, D.J. Poliovirus cis-acting replication element-dependent VPg Uridylylation lowers the Km of the initiating nucleoside triphosphate for viral RNA replication. J. Virol. 2008, 82, 9400–9408. [Google Scholar] [CrossRef]
- Ambros, V.; Baltimore, D. Protein is linked to the 5’ end of poliovirus RNA by a phosphodiester linkage to tyrosine. J. Biol. Chem. 1978, 253, 5263–5266. [Google Scholar] [PubMed]
- Fernandez-Munoz, R.; Darnell, J.E. Structural difference between the 5’ termini of viral and cellular mRNA in poliovirus-infected cells: Possible basis for the inhibition of host protein synthesis. J. Virol. 1976, 18, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, A.; Kitamura, N.; Golini, F.; Wimmer, E. The 5’-terminal structures of poliovirion RNA and poliovirus mRNA differ only in the genome-linked protein VPg. Proc. Natl. Acad. Sci. USA 1977, 74, 5345–5349. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, A.; Lee, Y.F.; Wimmer, E. The 5’ end of poliovirus mRNA is not capped with m7G(5’)ppp(5’)Np. Proc. Natl. Acad. Sci. USA 1976, 73, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Leveque, N.; Semler, B.L. A 21st century perspective of poliovirus replication. PLoS Pathog. 2015, 11, e1004825. [Google Scholar] [CrossRef]
- Ambros, V.; Pettersson, R.F.; Baltimore, D. An enzymatic activity in uninfected cells that cleaves the linkage between poliovirion RNA and the 5’ terminal protein. Cell 1978, 15, 1439–1446. [Google Scholar] [CrossRef]
- Virgen-Slane, R.; Rozovics, J.M.; Fitzgerald, K.D.; Ngo, T.; Chou, W.; van der Heden van Noort, G.J.; Filippov, D.V.; Gershon, P.D.; Semler, B.L. An RNA virus hijacks an incognito function of a DNA repair enzyme. Proc. Natl. Acad. Sci. USA 2012, 109, 14634–14639. [Google Scholar] [CrossRef]
- Cortes Ledesma, F.; El Khamisy, S.F.; Zuma, M.C.; Osborn, K.; Caldecott, K.W. A human 5’-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 2009, 461, 674–678. [Google Scholar] [CrossRef]
- Maciejewski, S.; Nguyen, J.H.; Gomez-Herreros, F.; Cortes-Ledesma, F.; Caldecott, K.W.; Semler, B.L. Divergent Requirement for a DNA Repair Enzyme during Enterovirus Infections. MBio 2015, 7, e01931-01915. [Google Scholar] [CrossRef]
- Langereis, M.A.; Feng, Q.; Nelissen, F.H.; Virgen-Slane, R.; van der Heden van Noort, G.J.; Maciejewski, S.; Filippov, D.V.; Semler, B.L.; van Delft, F.L.; van Kuppeveld, F.J. Modification of picornavirus genomic RNA using ’click’ chemistry shows that unlinking of the VPg peptide is dispensable for translation and replication of the incoming viral RNA. Nucleic Acids Res. 2014, 42, 2473–2482. [Google Scholar] [CrossRef]
- Nugent, C.I.; Johnson, K.L.; Sarnow, P.; Kirkegaard, K. Functional coupling between replication and packaging of poliovirus replicon RNA. J. Virol. 1999, 73, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Golini, F.; Semler, B.L.; Dorner, A.J.; Wimmer, E. Protein-linked RNA of poliovirus is competent to form an initiation complex of translation in vitro. Nature 1980, 287, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Andino, R.; Rieckhof, G.E.; Baltimore, D. A functional ribonucleoprotein complex forms around the 5’ end of poliovirus RNA. Cell 1990, 63, 369–380. [Google Scholar] [CrossRef]
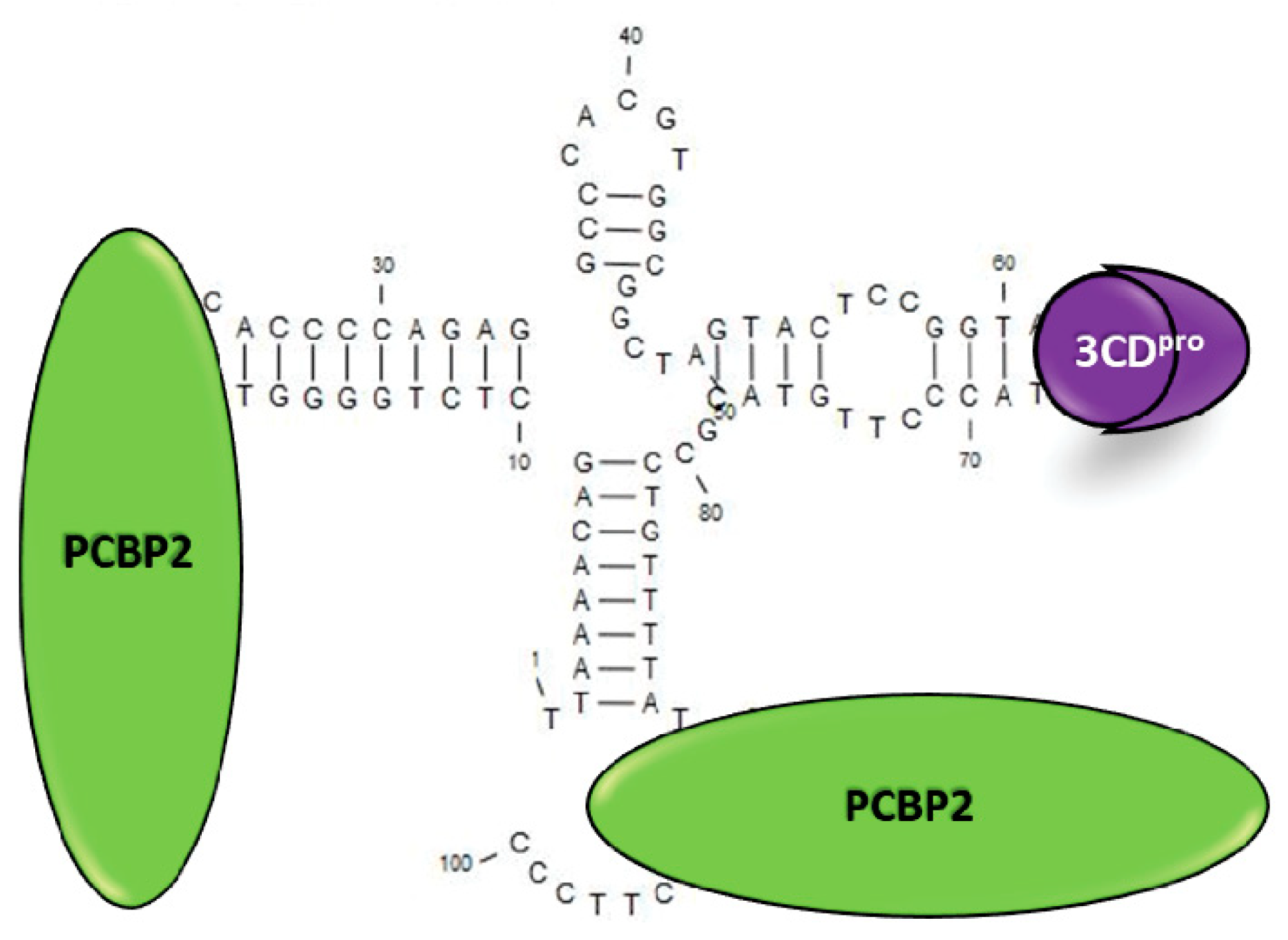
- Parsley, T.B.; Towner, J.S.; Blyn, L.B.; Ehrenfeld, E.; Semler, B.L. Poly (rC) binding protein 2 forms a ternary complex with the 5’-terminal sequences of poliovirus RNA and the viral 3CD proteinase. RNA 1997, 3, 1124–1134. [Google Scholar]
- Gamarnik, A.V.; Andino, R. Two functional complexes formed by KH domain containing proteins with the 5’ noncoding region of poliovirus RNA. RNA 1997, 3, 882–892. [Google Scholar]
- Toyoda, H.; Franco, D.; Fujita, K.; Paul, A.V.; Wimmer, E. Replication of poliovirus requires binding of the poly(rC) binding protein to the cloverleaf as well as to the adjacent C-rich spacer sequence between the cloverleaf and the internal ribosomal entry site. J. Virol. 2007, 81, 10017–10028. [Google Scholar] [CrossRef]
- Herold, J.; Andino, R. Poliovirus RNA replication requires genome circularization through a protein-protein bridge. Mol. Cell 2001, 7, 581–591. [Google Scholar] [CrossRef]
- Barton, D.J.; O’Donnell, B.J.; Flanegan, J.B. 5’ cloverleaf in poliovirus RNA is a cis-acting replication element required for negative-strand synthesis. EMBO J. 2001, 20, 1439–1448. [Google Scholar] [CrossRef]
- Vogt, D.A.; Andino, R. An RNA element at the 5’-end of the poliovirus genome functions as a general promoter for RNA synthesis. PLoS Pathog. 2010, 6, e1000936. [Google Scholar] [CrossRef]
- Kempf, B.J.; Barton, D.J. Poly(rC) binding proteins and the 5’ cloverleaf of uncapped poliovirus mRNA function during de novo assembly of polysomes. J. Virol. 2008, 82, 5835–5846. [Google Scholar] [CrossRef]
- Murray, K.E.; Roberts, A.W.; Barton, D.J. Poly(rC) binding proteins mediate poliovirus mRNA stability. RNA 2001, 7, 1126–1141. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Ogram, S.A.; Morasco, B.J.; Spear, A.; Chapman, N.M.; Flanegan, J.B. Functional role of the 5’ terminal cloverleaf in Coxsackievirus RNA replication. Virology 2009, 393, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Ullmer, W.; Semler, B.L. Diverse Strategies Used by Picornaviruses to Escape Host RNA Decay Pathways. Viruses 2016, 8, 335. [Google Scholar] [CrossRef] [PubMed]
- Haller, A.A.; Semler, B.L. Linker scanning mutagenesis of the internal ribosome entry site of poliovirus RNA. J. Virol. 1992, 66, 5075–5086. [Google Scholar] [CrossRef] [PubMed]
- Herold, J.; Andino, R. Poliovirus requires a precise 5’ end for efficient positive-strand RNA synthesis. J. Virol. 2000, 74, 6394–6400. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Kuhn, R.J.; Tada, H.; Ypma-Wong, M.F.; Semler, B.L.; Wimmer, E. Mutational analysis of the genome-linked protein VPg of poliovirus. J. Virol. 1988, 62, 4207–4215. [Google Scholar] [CrossRef]
- Walter, B.L.; Parsley, T.B.; Ehrenfeld, E.; Semler, B.L. Distinct poly(rC) binding protein KH domain determinants for poliovirus translation initiation and viral RNA replication. J. Virol. 2002, 76, 12008–12022. [Google Scholar] [CrossRef]
- Asare, E.; Mugavero, J.; Jiang, P.; Wimmer, E.; Paul, A.V. A Single Amino Acid Substitution in Poliovirus Nonstructural Protein 2CATPase Causes Conditional Defects in Encapsidation and Uncoating. J. Virol. 2016, 90, 6174–6186. [Google Scholar] [CrossRef]
- Rozovics, J.M.; Virgen-Slane, R.; Semler, B.L. Engineered picornavirus VPg-RNA substrates: Analysis of a tyrosyl-RNA phosphodiesterase activity. PLoS ONE 2011, 6, e16559. [Google Scholar] [CrossRef]
- Fitzgerald, K.D.; Semler, B.L. Re-localization of cellular protein SRp20 during poliovirus infection: Bridging a viral IRES to the host cell translation apparatus. PLoS Pathog. 2011, 7, e1002127. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Alvarez, E.; Carrasco, L. The multifaceted poliovirus 2A protease: Regulation of gene expression by picornavirus proteases. J. Biomed. Biotechnol. 2011, 2011, 369648. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, G.L.; Benedicto, I.; Philp, N.J.; Rodriguez-Boulan, E. Plasma membrane protein polarity and trafficking in RPE cells: Past, present and future. Exp. Eye Res. 2014, 126, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Chen, R.; Teesalu, T.; Ruoslahti, E.; Clegg, D.O. Reprogramming human retinal pigmented epithelial cells to neurons using recombinant proteins. Stem Cells Transl. Med. 2014, 3, 1526–1534. [Google Scholar] [CrossRef]
- Wang, S.Z.; Ma, W.; Yan, R.T.; Mao, W. Generating retinal neurons by reprogramming retinal pigment epithelial cells. Expert Opin. Biol. Ther. 2010, 10, 1227–1239. [Google Scholar] [CrossRef]
- Gao, R.; Schellenberg, M.J.; Huang, S.Y.; Abdelmalak, M.; Marchand, C.; Nitiss, K.C.; Nitiss, J.L.; Williams, R.S.; Pommier, Y. Proteolytic degradation of topoisomerase II (Top2) enables the processing of Top2.DNA and Top2.RNA covalent complexes by tyrosyl-DNA-phosphodiesterase 2 (TDP2). J. Biol. Chem. 2014, 289, 17960–17969. [Google Scholar] [CrossRef]
- Gradi, A.; Svitkin, Y.V.; Imataka, H.; Sonenberg, N. Proteolysis of human eukaryotic translation initiation factor eIF4GII, but not eIF4GI, coincides with the shutoff of host protein synthesis after poliovirus infection. Proc. Natl. Acad. Sci. USA 1998, 95, 11089–11094. [Google Scholar] [CrossRef]
- Ventoso, I.; MacMillan, S.E.; Hershey, J.W.; Carrasco, L. Poliovirus 2A proteinase cleaves directly the eIF-4G subunit of eIF-4F complex. FEBS Lett. 1998, 435, 79–83. [Google Scholar] [CrossRef]
- Kametani, Y.; Takahata, C.; Narita, T.; Tanaka, K.; Iwai, S.; Kuraoka, I. FEN1 participates in repair of the 5’-phosphotyrosyl terminus of DNA single-strand breaks. Carcinogenesis 2016, 37, 56–62. [Google Scholar] [CrossRef]
- Hoa, N.N.; Shimizu, T.; Zhou, Z.W.; Wang, Z.Q.; Deshpande, R.A.; Paull, T.T.; Akter, S.; Tsuda, M.; Furuta, R.; Tsutsui, K.; et al. Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes. Mol Cell 2016, 64, 580–592. [Google Scholar] [CrossRef]
- Li, C.; Sun, S.Y.; Khuri, F.R.; Li, R. Pleiotropic functions of EAPII/TTRAP/TDP2: Cancer development, chemoresistance and beyond. Cell Cycle 2011, 10, 3274–3283. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Fan, S.; Owonikoko, T.K.; Khuri, F.R.; Sun, S.Y.; Li, R. Oncogenic role of EAPII in lung cancer development and its activation of the MAPK-ERK pathway. Oncogene 2011, 30, 3802–3812. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lim, B.K.; Nam, J.H.; Gil, C.O.; Yun, S.H.; Choi, J.H.; Kim, D.K.; Jeon, E.S. Coxsackievirus B3 replication is related to activation of the late extracellular signal-regulated kinase (ERK) signal. Virus Res. 2005, 113, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Yanagawa, B.; Zhang, J.; Luo, Z.; Zhang, M.; Esfandiarei, M.; Carthy, C.; Wilson, J.E.; Yang, D.; McManus, B.M. Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. J. Virol. 2002, 76, 3365–3373. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, S.; Chen, Y.; Wang, T.; Dong, C.; Wo, X.; Zhang, J.; Dong, Y.; Xu, W.; Feng, X.; et al. The Capsid Protein VP1 of Coxsackievirus B Induces Cell Cycle Arrest by Up-Regulating Heat Shock Protein 70. Front. Microbiol. 2019, 10, 1633. [Google Scholar] [CrossRef]
- Pype, S.; Declercq, W.; Ibrahimi, A.; Michiels, C.; Van Rietschoten, J.G.; Dewulf, N.; de Boer, M.; Vandenabeele, P.; Huylebroeck, D.; Remacle, J.E. TTRAP, a novel protein that associates with CD40, tumor necrosis factor (TNF) receptor-75 and TNF receptor-associated factors (TRAFs), and that inhibits nuclear factor-kappa B activation. J. Biol. Chem. 2000, 275, 18586–18593. [Google Scholar] [CrossRef]
- Neznanov, N.; Chumakov, K.M.; Neznanova, L.; Almasan, A.; Banerjee, A.K.; Gudkov, A.V. Proteolytic cleavage of the p65-RelA subunit of NF-kappaB during poliovirus infection. J. Biol. Chem. 2005, 280, 24153–24158. [Google Scholar] [CrossRef]
- Li, Q.; Zheng, Z.; Liu, Y.; Zhang, Z.; Liu, Q.; Meng, J.; Ke, X.; Hu, Q.; Wang, H. 2C Proteins of Enteroviruses Suppress IKKbeta Phosphorylation by Recruiting Protein Phosphatase 1. J. Virol. 2016, 90, 5141–5151. [Google Scholar] [CrossRef]
- Franco, D.; Pathak, H.B.; Cameron, C.E.; Rombaut, B.; Wimmer, E.; Paul, A.V. Stimulation of poliovirus RNA synthesis and virus maturation in a HeLa cell-free in vitro translation-RNA replication system by viral protein 3CDpro. Virol. J. 2005, 2, 86. [Google Scholar] [CrossRef] [PubMed]
- Vance, L.M.; Moscufo, N.; Chow, M.; Heinz, B.A. Poliovirus 2C region functions during encapsidation of viral RNA. J. Virol. 1997, 71, 8759–8765. [Google Scholar] [CrossRef]
- Tijsma, A.; Thibaut, H.J.; Franco, D.; Dallmeier, K.; Neyts, J. Hydantoin: The mechanism of its in vitro anti-enterovirus activity revisited. Antiviral Res. 2016, 133, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Wang, G.; Yang, F.; Cao, W.; Mao, R.; Du, X.; Zhang, X.; Li, C.; Li, D.; Zhang, K.; et al. Foot-and-Mouth Disease Virus Viroporin 2B Antagonizes RIG-I-Mediated Antiviral Effects by Inhibition of Its Protein Expression. J. Virol. 2016, 90, 11106–11121. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zhang, Y.; Zou, J.; Yuan, Z.; Yi, Z. Replicase-mediated shielding of the poliovirus replicative double-stranded RNA to avoid recognition by MDA5. J. Gen. Virol. 2018, 99, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Bonin, M.; Oberstrass, J.; Lukacs, N.; Ewert, K.; Oesterschulze, E.; Kassing, R.; Nellen, W. Determination of preferential binding sites for anti-dsRNA antibodies on double-stranded RNA by scanning force microscopy. RNA 2000, 6, 563–570. [Google Scholar] [CrossRef]
- Ye, C.J.; Sharpe, Z.; Alemara, S.; Mackenzie, S.; Liu, G.; Abdallah, B.; Horne, S.; Regan, S.; Heng, H.H. Micronuclei and Genome Chaos: Changing the System Inheritance. Genes 2019, 10, 366. [Google Scholar] [CrossRef]
- Dougherty, J.D.; White, J.P.; Lloyd, R.E. Poliovirus-mediated disruption of cytoplasmic processing bodies. J. Virol. 2011, 85, 64–75. [Google Scholar] [CrossRef]
- Ren, X.; Linehan, M.M.; Iwasaki, A.; Pyle, A.M. RIG-I Selectively Discriminates against 5’-Monophosphate RNA. Cell Rep. 2019, 26, 2019–2027 e2014. [Google Scholar] [CrossRef]














© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holmes, A.C.; Zagnoli-Vieira, G.; Caldecott, K.W.; Semler, B.L. Effects of TDP2/VPg Unlinkase Activity on Picornavirus Infections Downstream of Virus Translation. Viruses 2020, 12, 166. https://doi.org/10.3390/v12020166
Holmes AC, Zagnoli-Vieira G, Caldecott KW, Semler BL. Effects of TDP2/VPg Unlinkase Activity on Picornavirus Infections Downstream of Virus Translation. Viruses. 2020; 12(2):166. https://doi.org/10.3390/v12020166
Chicago/Turabian StyleHolmes, Autumn C., Guido Zagnoli-Vieira, Keith W. Caldecott, and Bert L. Semler. 2020. "Effects of TDP2/VPg Unlinkase Activity on Picornavirus Infections Downstream of Virus Translation" Viruses 12, no. 2: 166. https://doi.org/10.3390/v12020166
APA StyleHolmes, A. C., Zagnoli-Vieira, G., Caldecott, K. W., & Semler, B. L. (2020). Effects of TDP2/VPg Unlinkase Activity on Picornavirus Infections Downstream of Virus Translation. Viruses, 12(2), 166. https://doi.org/10.3390/v12020166