Diversity and Host Interactions among Virulent and Temperate Baltic Sea Flavobacterium Phages

 ,
,  , and
, and 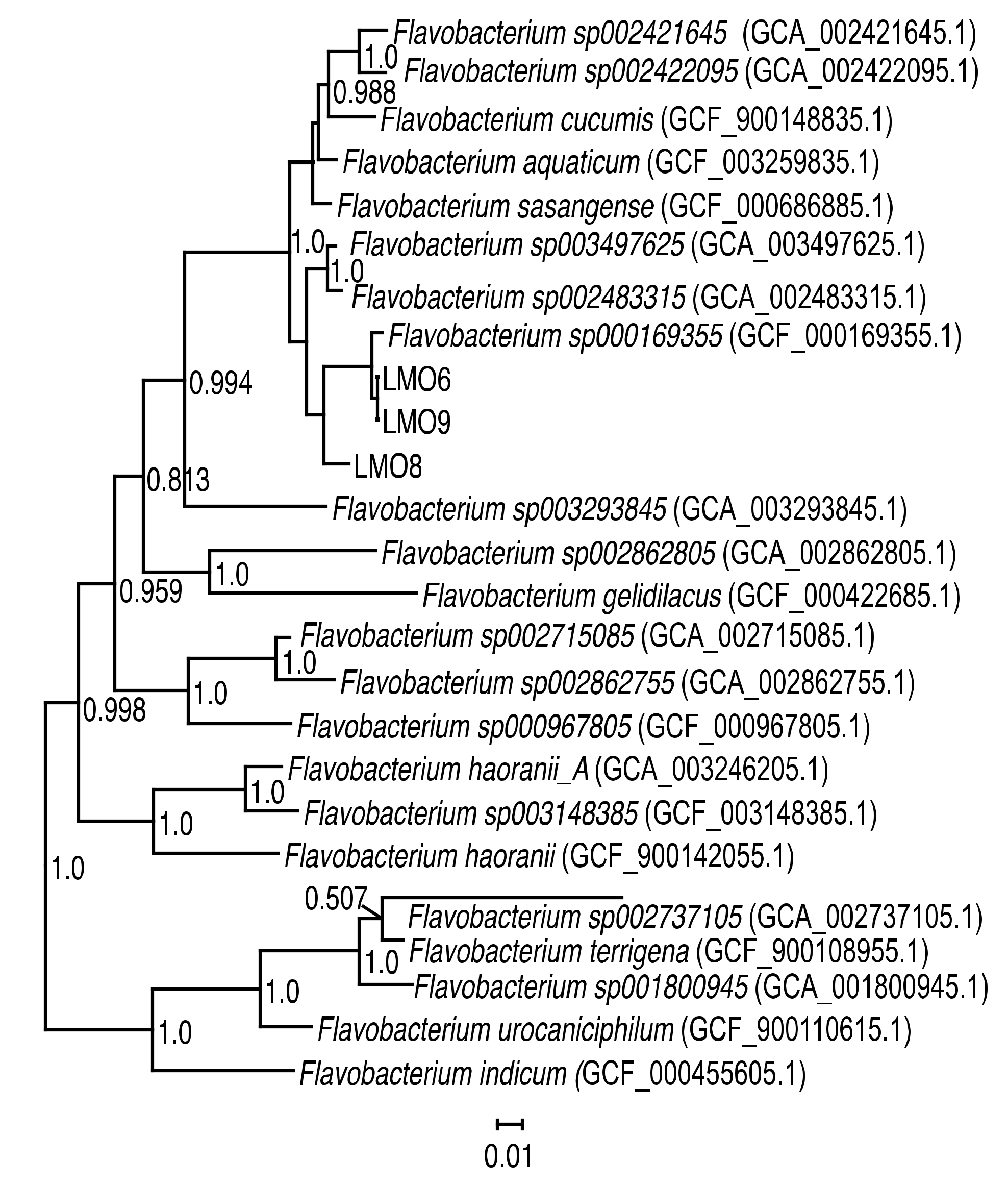{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Isolation and DNA Extraction
2.2. Viral Isolation
2.3. Host Range Investigation
2.4. Prophage Induction Experiments with LMO6
2.5. Morphological Examination Using Transmission Electron Microscopy (TEM)
2.6. Viral DNA Extraction
2.7. Sequencing of Viral and Bacterial Genomes
2.8. Assembly, Annotation, and Pan-Genome Analysis
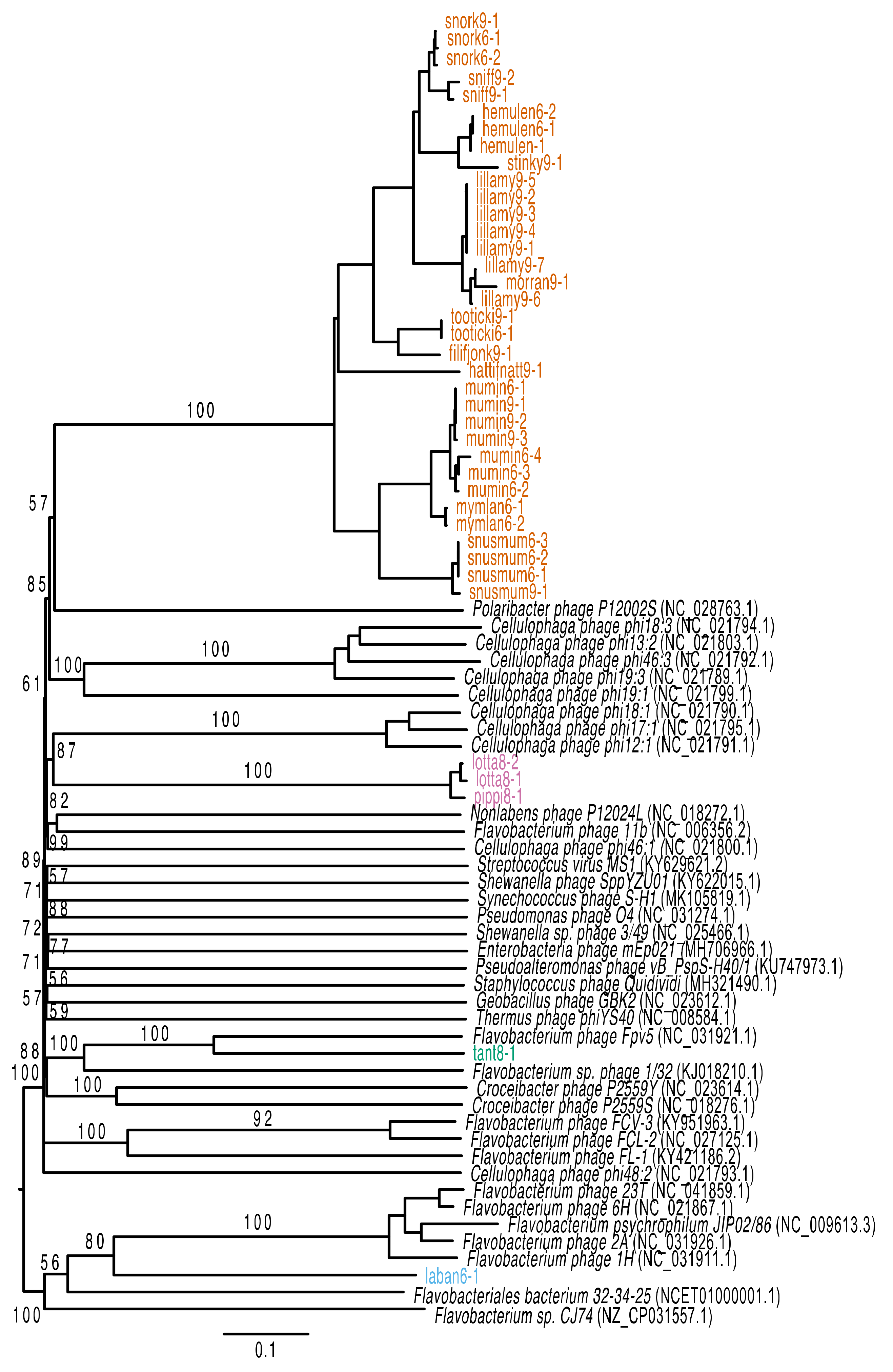
2.9. Phylogenetic Analysis
2.10. Mapping of Environmental Reads to the Phage Genomes and Host Bacteria
2.11. Mapping of Reads to Confirm Prophage Induction
2.12. Accession Numbers
3. Results
3.1. Characteristics of Flavobacterium spp. Strains
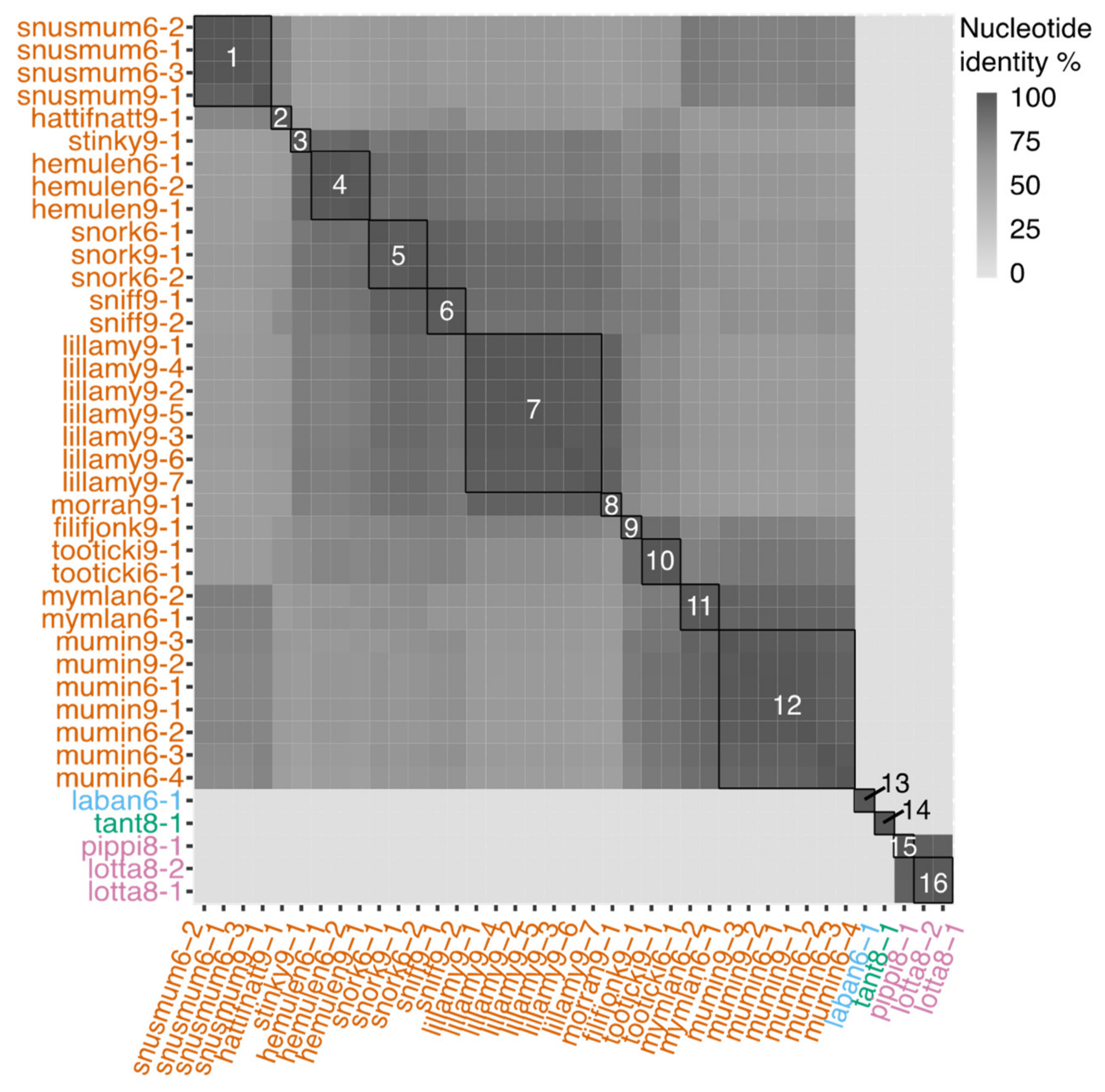
3.2. Genomic Characteristics of Lytic Phages
3.3. Genomic Characteristics of the Temperate Phage
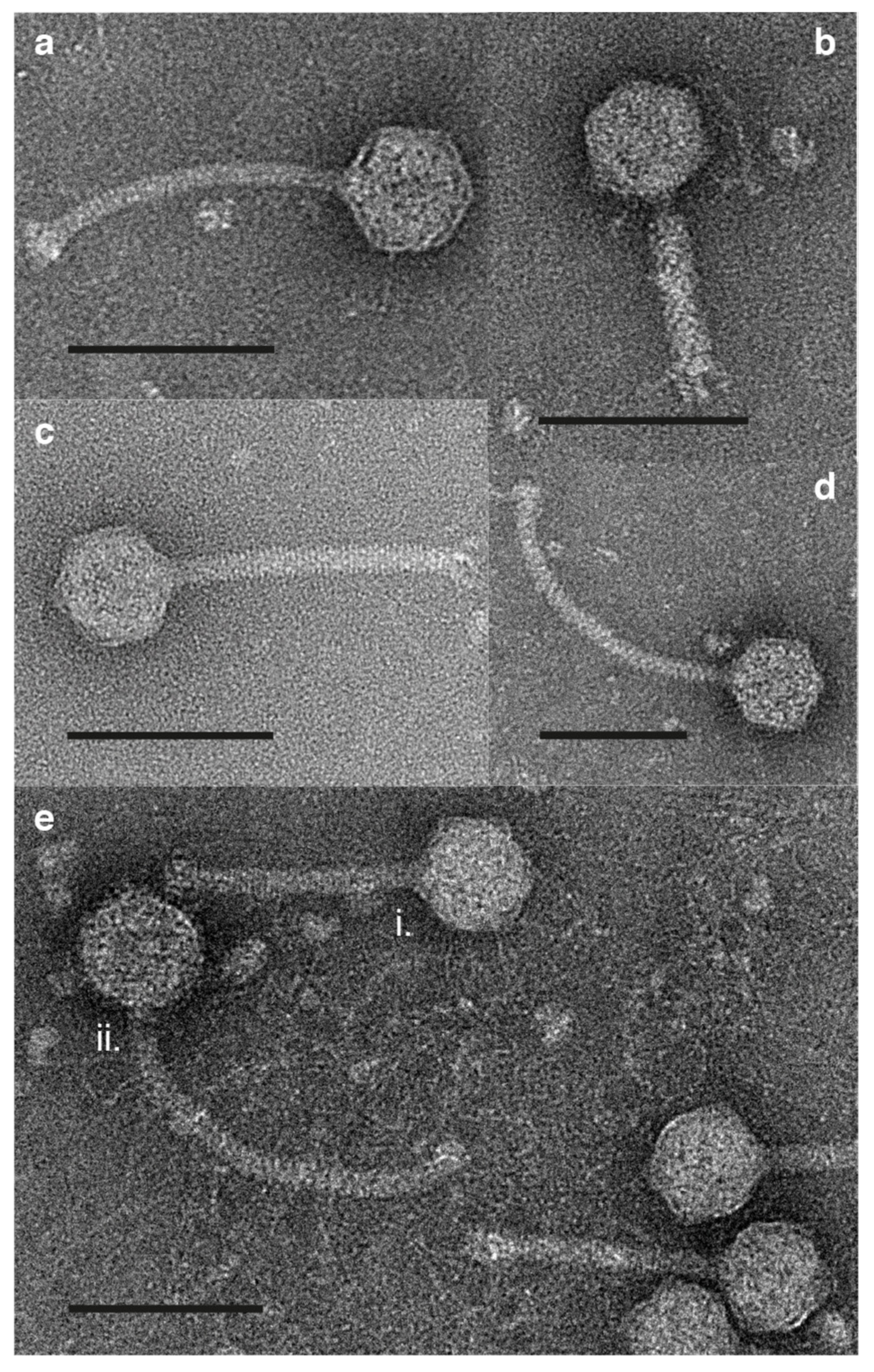
3.4. Morphology
3.5. Host Range
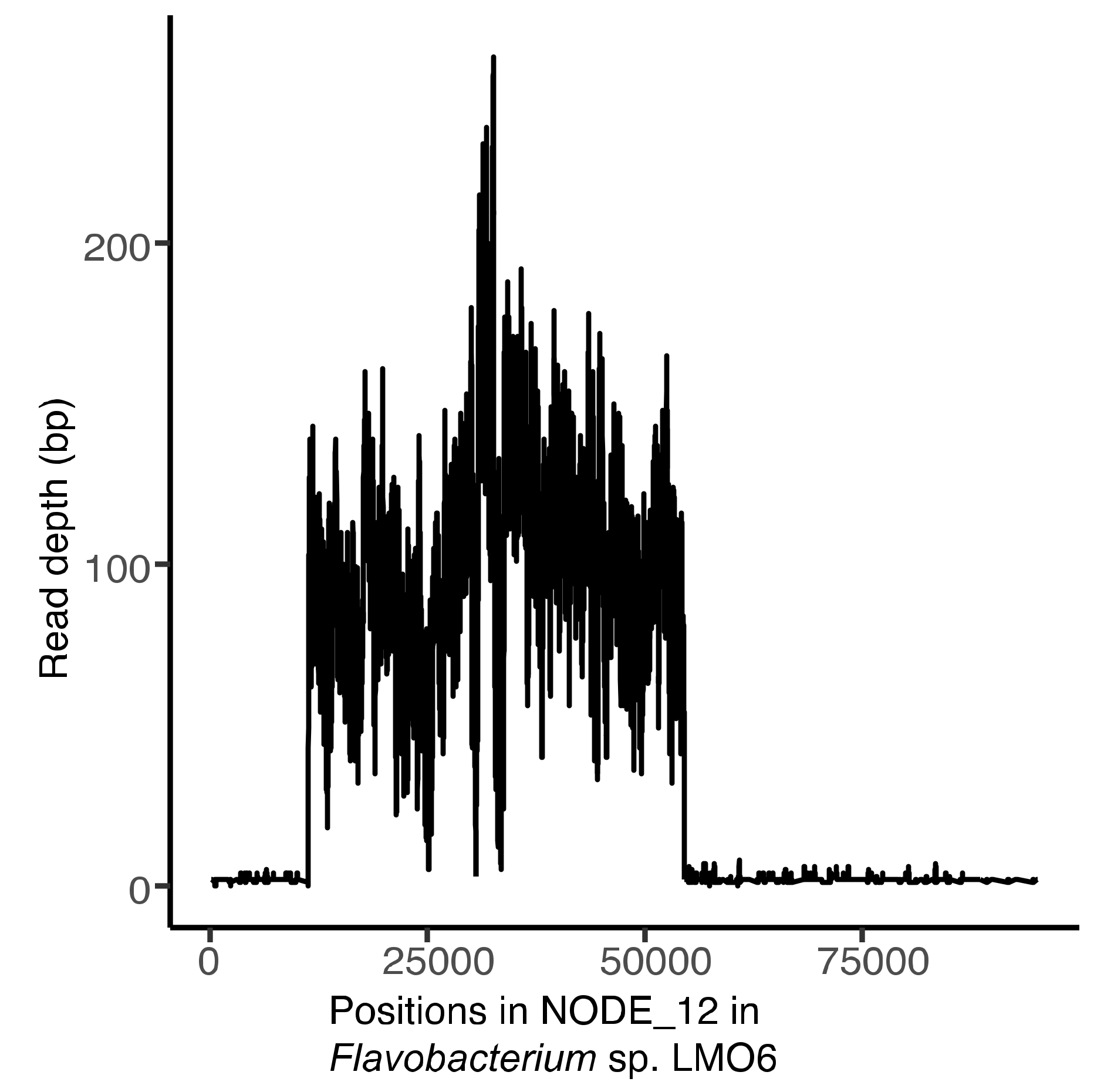
3.6. Induction of laban6-1 From LMO6
3.7. Temporal and Spatial Variation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bergh, O.; Borsheim, K.Y.; Bratbak, G.; Heldal, M. High abundance of viruses found in aquatic environments. Nature 1989, 340, 467–468. [Google Scholar] [CrossRef]
- Suttle, C.A. Marine viruses-major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Weitz, J.S.; Poisot, T.; Meyer, J.R.; Flores, C.O.; Valverde, S.; Sullivan, M.B.; Hochberg, M.E. Phage-bacteria infection networks. Trends Microbiol. 2013, 21, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Holmfeldt, K.; Solonenko, N.; Howard-Varona, C.; Moreno, M.; Malmstrom, R.R.; Blow, M.J.; Sullivan, M.B. Large-scale maps of variable infection efficiencies in aquatic Bacteroidetes phage-host model systems. Environ. Microbiol. 2016, 18, 3949–3961. [Google Scholar] [CrossRef] [PubMed]
- Mann, N.H.; Cook, A.; Millard, A.; Bailey, S.; Clokie, M. Marine ecosystems: Bacterial photosynthesis genes in a virus. Nature 2003, 424, 741. [Google Scholar] [CrossRef]
- Lindell, D.; Jaffe, J.D.; Coleman, M.L.; Futschik, M.E.; Axmann, I.M.; Rector, T.; Kettler, G.; Sullivan, M.B.; Steen, R.; Hess, W.R.; et al. Genome-wide expression dynamics of a marine virus and host reveal features of co-evolution. Nature 2007, 449, 83–86. [Google Scholar] [CrossRef]
- Breitbart, M.; Bonnain, C.; Malki, K.; Sawaya, N.A. Phage puppet masters of the marine microbial realm. Nat. Microbiol. 2018, 3, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Middelboe, M.; Holmfeldt, K.; Riemann, L.; Nybroe, O.; Haaber, J. Bacteriophages drive strain diversification in a marine Flavobacterium: Implications for phage resistance and physiological properties. Environ. Microbiol. 2009, 11, 1971–1982. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, B.L.; Hallam, S.J.; Sullivan, M.B. Metabolic reprogramming by viruses in the sunlit and dark ocean. Genome Biol. 2013, 14, R123. [Google Scholar] [CrossRef]
- Howard-Varona, C.; Hargreaves, K.R.; Solonenko, N.E.; Markillie, L.M.; White, R.A.; Brewer, H.M.; Ansong, C.; Orr, G.; Adkins, J.N.; Sullivan, M.B. Multiple mechanisms drive phage infection efficiency in nearly identical hosts. ISME J. 2018, 12, 1605–1618. [Google Scholar] [CrossRef]
- Levin, B.; Lenski, R. Coevolution in bacteria and their viruses and plasmids. In Coevolution; Futuyama, D.a.S.M., Ed.; Sinauer Associates: Sunderland, MA, USA, 1983; pp. 99–127. [Google Scholar]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Bratbak, G.; Thingstad, F.; Heldal, M. Viruses and the microbial loop. Microb. Ecol. 1994, 28, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Warwick-Dugdale, J.; Buchholz, H.H.; Allen, M.J.; Temperton, B. Host-hijacking and planktonic piracy: How phages command the microbial high seas. Virol. J. 2019, 16. [Google Scholar] [CrossRef] [PubMed]
- Waldor, M.K.; Mekalanos, J.J. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 1996, 272, 1910–1914. [Google Scholar] [CrossRef]
- Chen, Y.; Golding, I.; Sawai, S.; Guo, L.; Cox, E.C. Population fitness and the regulation of Escherichia coli genes by bacterial viruses. PLoS Biol. 2005, 3, e229. [Google Scholar] [CrossRef]
- Paul, J.H. Prophages in marine bacteria: Dangerous molecular time bombs or the key to survival in the seas? ISME J. 2008, 2, 579–589. [Google Scholar] [CrossRef]
- Jiang, S.C.; Paul, J.H. Seasonal and diel abundance of viruses and occurrence of lysogeny/bacteriocinogeny in the marine environment. Mar. Ecol. Prog. Ser. 1994, 104, 163–172. [Google Scholar] [CrossRef]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. eLife 2015, 4, e08490. [Google Scholar] [CrossRef]
- Cochran, P.K.; Paul, J.H. Seasonal abundance of lysogenic bacteria in a subtropical estuary. Appl. Environ. Microb. 1998, 64, 2308–2312. [Google Scholar] [CrossRef]
- Williamson, S.J.; Houchin, L.A.; McDaniel, L.; Paul, J.H. Seasonal variation in lysogeny as depicted by prophage induction in Tampa Bay, Florida. Appl. Environ. Microb. 2002, 68, 4307–4314. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Hurwitz, B.L.; Schofield, O.; Ducklow, H.W.; Sullivan, M.B. Seasonal time bombs: Dominant temperate viruses affect Southern Ocean microbial dynamics. ISME J. 2016, 10, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Chuvochina, M.S.; Marie, D.; Chevaillier, S.; Petit, J.R.; Normand, P.; Alekhina, I.A.; Bulat, S.A. Community variability of bacteria in alpine snow (Mont Blanc) containing Saharan dust deposition and their snow colonisation potential. Microbes Environ. 2011, 26, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.Z.; Allen, E.E.; Badger, J.H.; McCrow, J.P.; Paulsen, I.T.; Elbourne, L.D.H.; Thiagarajan, M.; Rusch, D.B.; Nealson, K.H.; Williamson, S.J.; et al. Influence of nutrients and currents on the genomic composition of microbes across an upwelling mosaic. ISME J. 2012, 6, 1403–1414. [Google Scholar] [CrossRef]
- Kirchman, D.L. The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 2002, 39, 91–100. [Google Scholar] [CrossRef]
- Pinhassi, J.; Sala, M.M.; Havskum, H.; Peters, F.; Guadayol, O.; Malits, A.; Marrase, C. Changes in bacterioplankton composition under different phytoplankton regimens. Appl. Environ. Microbiol. 2004, 70, 6753–6766. [Google Scholar] [CrossRef]
- Fernandez-Gomez, B.; Richter, M.; Schuler, M.; Pinhassi, J.; Acinas, S.G.; Gonzalez, J.M.; Pedros-Alio, C. Ecology of marine Bacteroidetes: A comparative genomics approach. ISME J. 2013, 7, 1026–1037. [Google Scholar] [CrossRef]
- Rodrigues, R.A.L.; Andrade, A.; Boratto, P.V.M.; Trindade, G.S.; Kroon, E.G.; Abrahao, J.S. An anthropocentric view of the virosphere-host relationship. Front. Microbiol. 2017, 8, 1673. [Google Scholar] [CrossRef]
- McBride, M.J. The family Flavobacteriaceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 643–676. [Google Scholar]
- Borriss, M.; Lombardot, T.; Glockner, F.O.; Becher, D.; Albrecht, D.; Schweder, T. Genome and proteome characterization of the psychrophilic Flavobacterium bacteriophage 11b. Extremophiles 2007, 11, 95–104. [Google Scholar] [CrossRef]
- Kang, I.; Jang, H.; Cho, J.C. Complete genome sequences of two Persicivirga bacteriophages, P12024S and P12024L. J. Virol. 2012, 86, 8907–8908. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Kang, D.; Cho, J.C. Complete genome sequence of Croceibacter bacteriophage P2559S. J. Virol. 2012, 86, 8912–8913. [Google Scholar] [CrossRef] [PubMed]
- Laanto, E.; Mäntynen, S.; De Colibus, L.; Marjakangas, J.; Gillum, A.; Stuart, D.I.; Ravantti, J.J.; Huiskonen, J.T.; Sundberg, L.R. Virus found in a boreal lake links ssDNA and dsDNA viruses. Proc. Natl. Acad. Sci. USA 2017, 114, 8378–8383. [Google Scholar] [CrossRef] [PubMed]
- Holmfeldt, K.; Middelboe, M.; Nybroe, O.; Riemann, L. Large variabilities in host strain susceptibility and phage host range govern interactions between lytic marine phages and their Flavobacterium hosts. Appl. Environ. Microbiol. 2007, 73, 6730–6739. [Google Scholar] [CrossRef] [PubMed]
- Holmfeldt, K.; Solonenko, N.; Shah, M.; Corrier, K.; Riemann, L.; Verberkmoes, N.C.; Sullivan, M.B. Twelve previously unknown phage genera are ubiquitous in global oceans. Proc. Natl. Acad. Sci. USA 2013, 110, 12798–12803. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Noriko, C.W.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef]
- Yutin, N.; Makarova, K.S.; Gussow, A.B.; Krupovic, M.; Segall, A.; Edwards, R.A.; Koonin, E.V. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol. 2018, 3, 38–46. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Khokhlova, E.V.; Fitzgerald, C.B.; Stockdale, S.R.; Draper, L.A.; Ross, R.P.; Hill, C. Phi CrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018, 9, 4781. [Google Scholar] [CrossRef]
- Riemann, L.; Grossart, H.P. Elevated lytic phage production as a consequence of particle colonization by a marine Flavobacterium (Cellulophaga sp.). Microb. Ecol. 2008, 56, 505–512. [Google Scholar] [CrossRef]
- Dang, V.T.; Howard-Varona, C.; Schwenck, S.; Sullivan, M.B. Variably lytic infection dynamics of large Bacteroidetes podovirus phi38:1 against two Cellulophaga baltica host strains. Environ. Microbiol. 2015, 17, 4659–4671. [Google Scholar] [CrossRef]
- Holmfeldt, K.; Howard-Varona, C.; Solonenko, N.; Sullivan, M.B. Contrasting genomic patterns and infection strategies of two co-existing Bacteroidetes podovirus genera. Environ. Microbiol. 2014, 16, 2501–2513. [Google Scholar] [CrossRef]
- Castillo, D.; Higuera, G.; Villa, M.; Middelboe, M.; Dalsgaard, I.; Madsen, L.; Espejo, R.T. Diversity of Flavobacterium psychrophilum and the potential use of its phages for protection against bacterial cold water disease in salmonids. J. Fish. Dis. 2012, 35, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, R.H.; Dalsgaard, I.; Middelboe, M.; Lauritsen, A.H.; Madsen, L. Detection and quantification of Flavobacterium psychrophilum-specific bacteriophages in vivo in rainbow trout upon oral administration: Implications for disease control in aquaculture. Appl. Environ. Microbiol. 2014, 80, 7683–7693. [Google Scholar] [CrossRef]
- Stenholm, A.R.; Dalsgaard, I.; Middelboe, M. Isolation and characterization of bacteriophages infecting the fish pathogen Flavobacterium psychrophilum. Appl. Environ. Microbiol. 2008, 74, 4070–4078. [Google Scholar] [CrossRef]
- Castillo, D.; Espejo, R.; Middelboe, M. Genomic structure of bacteriophage 6H and its distribution as prophage in Flavobacterium psychrophilum strains. FEMS Microbiol. Lett. 2014, 351, 51–58. [Google Scholar] [CrossRef]
- Castillo, D.; Middelboe, M. Genomic diversity of bacteriophages infecting the fish pathogen Flavobacterium psychrophilum. FEMS Microbiol. Lett. 2016, 363, fnw272. [Google Scholar] [CrossRef][Green Version]
- Duchaud, E.; Boussaha, M.; Loux, V.; Bernardet, J.F.; Michel, C.; Kerouault, B.; Mondot, S.; Nicolas, P.; Bossy, R.; Caron, C.; et al. Complete genome sequence of the fish pathogen Flavobacterium psychrophilum. Nat. Biotechnol. 2007, 25, 763–769. [Google Scholar] [CrossRef]
- Castillo, D.; Christiansen, R.H.; Dalsgaard, I.; Madsen, L.; Espejo, R.; Middelboe, M. Comparative genome analysis provides insights into the pathogenicity of Flavobacterium psychrophilum. PLoS ONE 2016, 11, e0152515. [Google Scholar] [CrossRef] [PubMed]
- Castillo, D.; Christiansen, R.H.; Dalsgaard, I.; Madsen, L.; Middelboe, M. Bacteriophage resistance mechanisms in the fish pathogen Flavobacterium psychrophilum: Linking genomic mutations to changes in bacterial virulence factors. Appl. Environ. Microbiol. 2015, 81, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.M.; Polz, M.F. Streamlining standard bacteriophage methods for higher throughput. MethodsX 2018, 5, 159–172. [Google Scholar] [CrossRef]
- Wickham, H. Tidyverse: Easily install and load ’tidyverse’ package; Version 1.3.0; RStudio Inc.: Boston, MA, USA, 2017. [Google Scholar]
- R Core Team. R: A language and environment for statistical computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- RStudio Team. RStudio: Integrated development for R; RStudio Inc.: Boston, MA, USA, 2015. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data; Version 0.7.2; The Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.M.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- McNair, K.; Zhou, C.; Dinsdale, E.A.; Souza, B.; Edwards, R.A. PHANOTATE: A novel approach to gene identification in phage genomes. Bioinformatics 2019, 35, 4537–4542. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Ågren, J.; Sundström, A.; Håfström, T.; Segerman, B. Gegenees: Fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PLoS ONE 2012, 7, e39107. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods UK 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.-A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. Selection of representative genomes for 24,706 bacterial and archaeal species clusters provide a complete genome-based taxonomy. bioRxiv 2019. [Google Scholar] [CrossRef]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Bio. 2012, 61, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef]
- Göker, M.; Garcia-Blázquez, G.; Voglmayr, H.; Tellería, M.T.; Martín, M.P. Molecular taxonomy of phytopathogenic fungi: A case study in Peronospora. PLoS ONE 2009, 4, e6319. [Google Scholar] [CrossRef]
- Nilsson, E.; Li, K.; Fridlund, J.; Šulčius, S.; Bunse, C.; Karlsson, C.M.G.; Lindh, M.; Lundin, D.; Pinhassi, J.; Holmfeldt, K. Genomic and seasonal variations among aquatic phages infecting the Baltic Sea gammaproteobacterium Rheinheimera sp. strain BAL341. Appl. Environ. Microbiol. 2019, 85, e01003–e01019. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Emerson, J.B.; Eloe-Fadrosh, E.A.; Sullivan, M.B. Benchmarking viromics: An in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 2017, 5, e3817. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Hugerth, L.W.; Larsson, J.; Alneberg, J.; Lindh, M.V.; Legrand, C.; Pinhassi, J.; Andersson, A.F. Metagenome-assembled genomes uncover a global brackish microbiome. Genome Biol. 2015, 16, 279. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Brister, J.R. How to name and classify your phage: An informal guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef]
- Kropinski, A.M.; Prangishvili, D.; Lavigne, R. Position paper: The creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environ. Microbiol. 2009, 11, 2775–2777. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Jacobs-Sera, D.; Lawrence, J.G.; Pope, W.H.; Russell, D.A.; Ko, C.C.; Weber, R.J.; Patel, M.C.; Germane, K.L.; Edgar, R.H.; et al. Comparative genomic analysis of 60 mycobacteriophage genomes: Genome clustering, gene acquisition, and gene size. J. Mol. Biol. 2010, 397, 119–143. [Google Scholar] [CrossRef]
- Sulcius, S.; Holmfeldt, K. Viruses of microorganisms in the Baltic Sea: Current state of research and perspectives. Mar. Biol. Res. 2016, 12, 115–124. [Google Scholar] [CrossRef]
- Luhtanen, A.M.; Eronen-Rasimus, E.; Kaartokallio, H.; Rintala, J.M.; Autio, R.; Roine, E. Isolation and characterization of phage-host systems from the Baltic Sea ice. Extremophiles 2014, 18, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Senkevich, T.G.; Dolja, V.V. The ancient virus world and evolution of cells. Biol. Direct. 2006, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Hawley, A.K.; Beltran, M.T.; Scofield, M.; Schwientek, P.; Stepanauskas, R.; Woyke, T.; Hallam, S.J.; Sullivan, M.B. Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta- genomics. eLife 2014, 3, e03125. [Google Scholar] [CrossRef]
- Breitbart, M. Marine viruses: Truth or dare. Ann. Rev. Mar. Sci. 2012, 4, 425–448. [Google Scholar] [CrossRef]
- Wang, X.; Kim, Y.; Ma, Q.; Hong, S.H.; Pokusaeva, K.; Sturino, J.M.; Wood, T.K. Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 2010, 1, 147. [Google Scholar] [CrossRef]
- Lwoff, A. Lysogeny. Bacteriol. Rev. 1953, 17, 269–337. [Google Scholar] [CrossRef]
- Alexeeva, S.; Martinez, J.A.G.; Spus, M.; Smid, E.J. Spontaneously induced prophages are abundant in a naturally evolved bacterial starter culture and deliver competitive advantage to the host. BMC Microbiol. 2018, 18. [Google Scholar] [CrossRef]
- Nanda, A.M.; Thormann, K.; Frunzke, J. Impact of spontaneous prophage induction on the fitness of bacterial populations and host-microbe interactions. J. Bacteriol. 2015, 197, 410–419. [Google Scholar] [CrossRef]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Jiang, S.C.; Paul, J.H. Occurrence of lysogenic bacteria in marine microbial communities as determined by prophage induction. Mar. Ecol. Prog. Ser. 1996, 142, 27–38. [Google Scholar] [CrossRef]
- Shan, J.; Korbsrisate, S.; Withatanung, P.; Adler, N.L.; Clokie, M.R.; Galyov, E.E. Temperature dependent bacteriophages of a tropical bacterial pathogen. Front. Microbiol. 2014, 5, 599. [Google Scholar] [CrossRef] [PubMed]
- Koskella, B.; Meaden, S. Understanding bacteriophage specificity in natural microbial communities. Viruses 2013, 5, 806–823. [Google Scholar] [CrossRef]
- Sullivan, M.B.; Waterbury, J.B.; Chisholm, S.W. Cyanophages infecting the oceanic cyanobacterium Prochlorococcus. Nature 2003, 424, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Arber, W. Host specificity of DNA produced by Escherichia coli: V. The role of methionine in the production of host specificity. J. Mol. Biol. 1965, 11, 247–256. [Google Scholar] [CrossRef]
- Hattman, S.; Fukasawa, T. Host-induced modification of T-even phages due to defective glucosylation of their DNA. Proc. Natl. Acad. Sci. USA 1963, 50, 297–300. [Google Scholar] [CrossRef]
- Luria, S.E. Host-induced modifications of viruses. Cold Spring Harb Symp Quant. Biol. 1953, 18, 237–244. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nilsson, E.; Bayfield, O.W.; Lundin, D.; Antson, A.A.; Holmfeldt, K. Diversity and Host Interactions among Virulent and Temperate Baltic Sea Flavobacterium Phages. Viruses 2020, 12, 158. https://doi.org/10.3390/v12020158
Nilsson E, Bayfield OW, Lundin D, Antson AA, Holmfeldt K. Diversity and Host Interactions among Virulent and Temperate Baltic Sea Flavobacterium Phages. Viruses. 2020; 12(2):158. https://doi.org/10.3390/v12020158
Chicago/Turabian StyleNilsson, Emelie, Oliver W. Bayfield, Daniel Lundin, Alfred A. Antson, and Karin Holmfeldt. 2020. "Diversity and Host Interactions among Virulent and Temperate Baltic Sea Flavobacterium Phages" Viruses 12, no. 2: 158. https://doi.org/10.3390/v12020158
APA StyleNilsson, E., Bayfield, O. W., Lundin, D., Antson, A. A., & Holmfeldt, K. (2020). Diversity and Host Interactions among Virulent and Temperate Baltic Sea Flavobacterium Phages. Viruses, 12(2), 158. https://doi.org/10.3390/v12020158