
Unveiling Viruses Associated with Gastroenteritis Using a Metagenomics Approach

 , ,
, , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Characterization
2.2. Viral Concentration, Free DNA Removal, Nucleic Acid Extraction, and Library Preamplification and Preparation Using Untargeted Viral Metagenomics (UVM) and Target Enrichment Sequencing (TES)
2.3. Bioinformatics Analysis
2.4. RT-PCR and RT-qPCR for Specific Viral Pathogens Sequencing
3. Results and Discussion
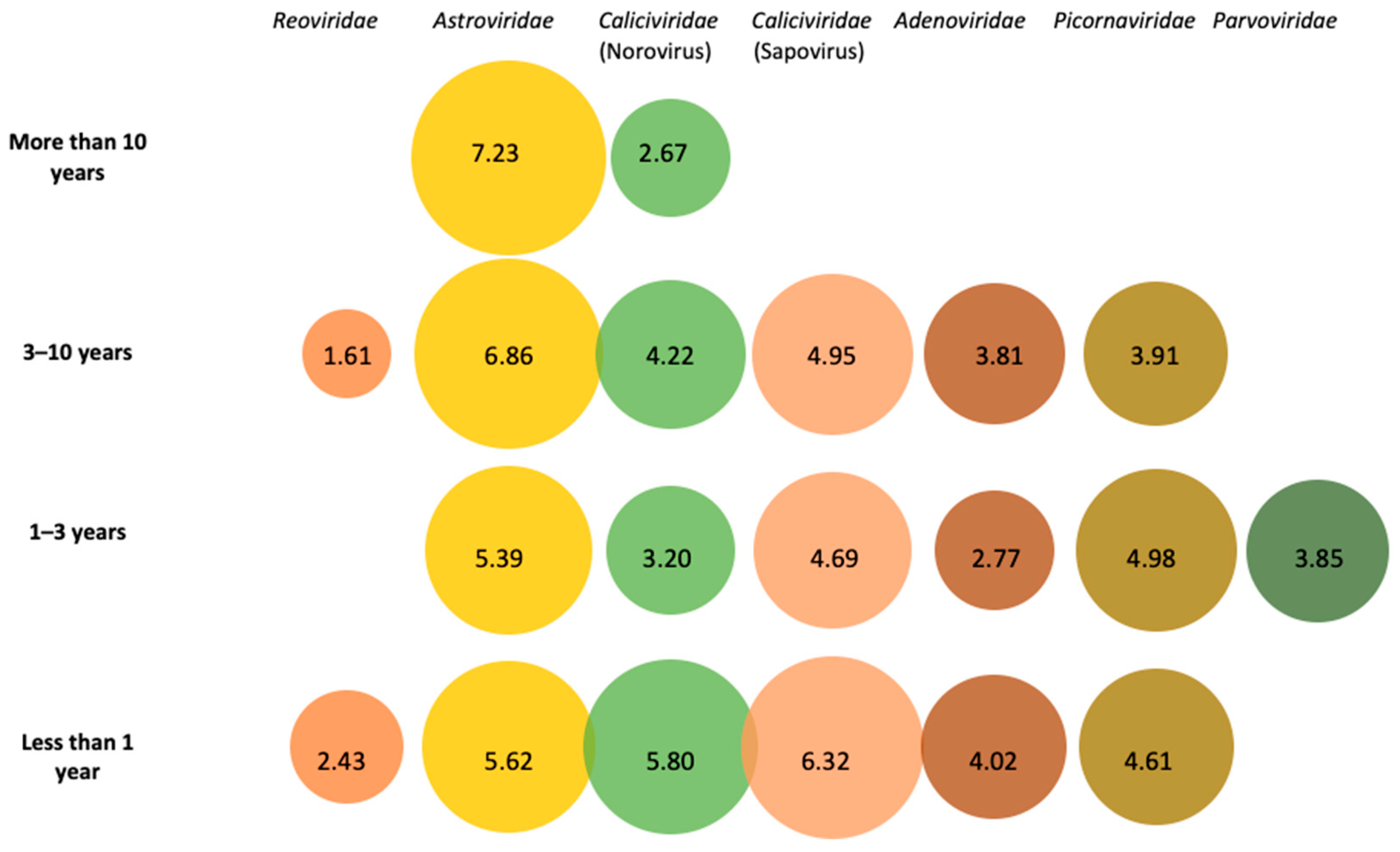
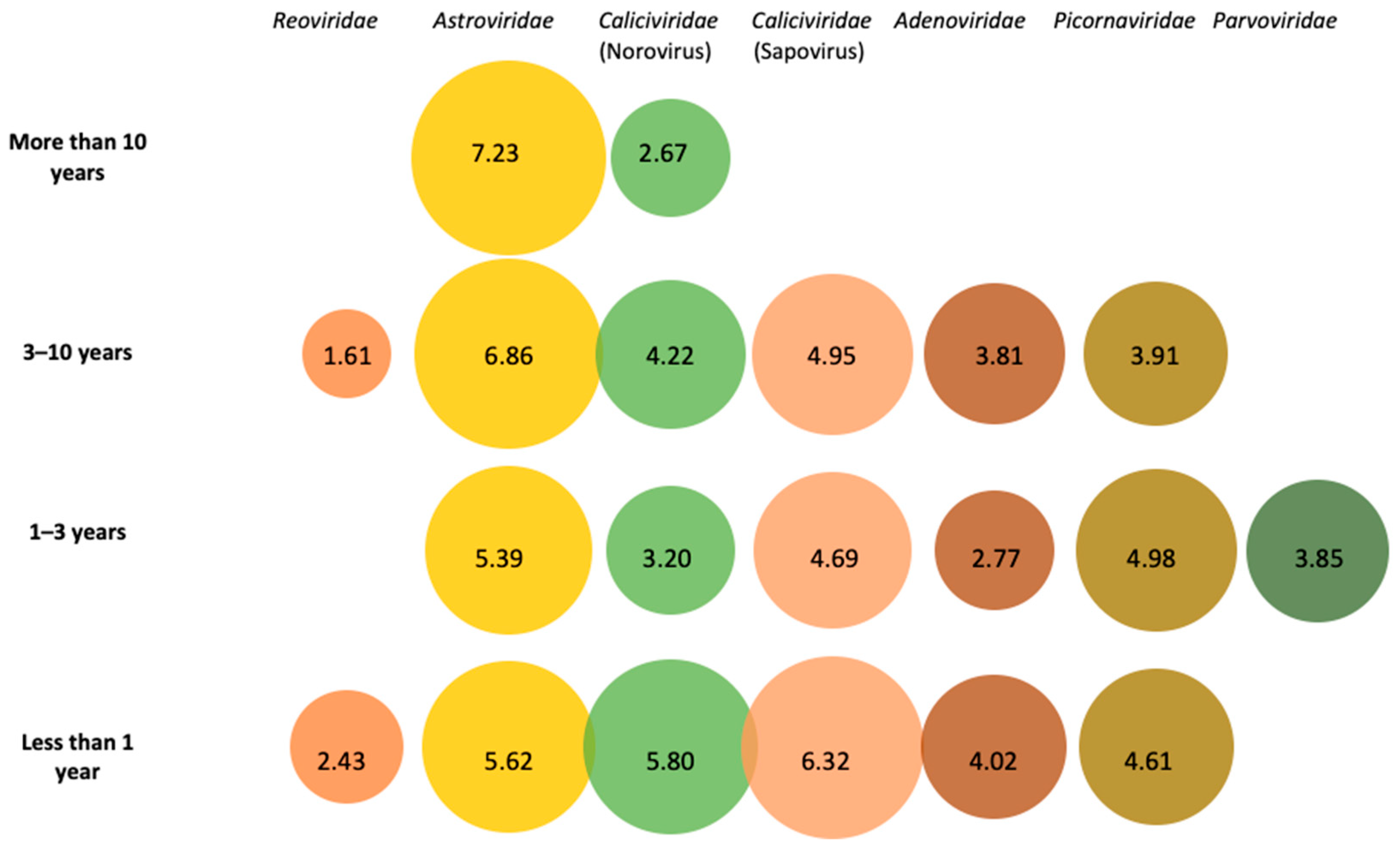
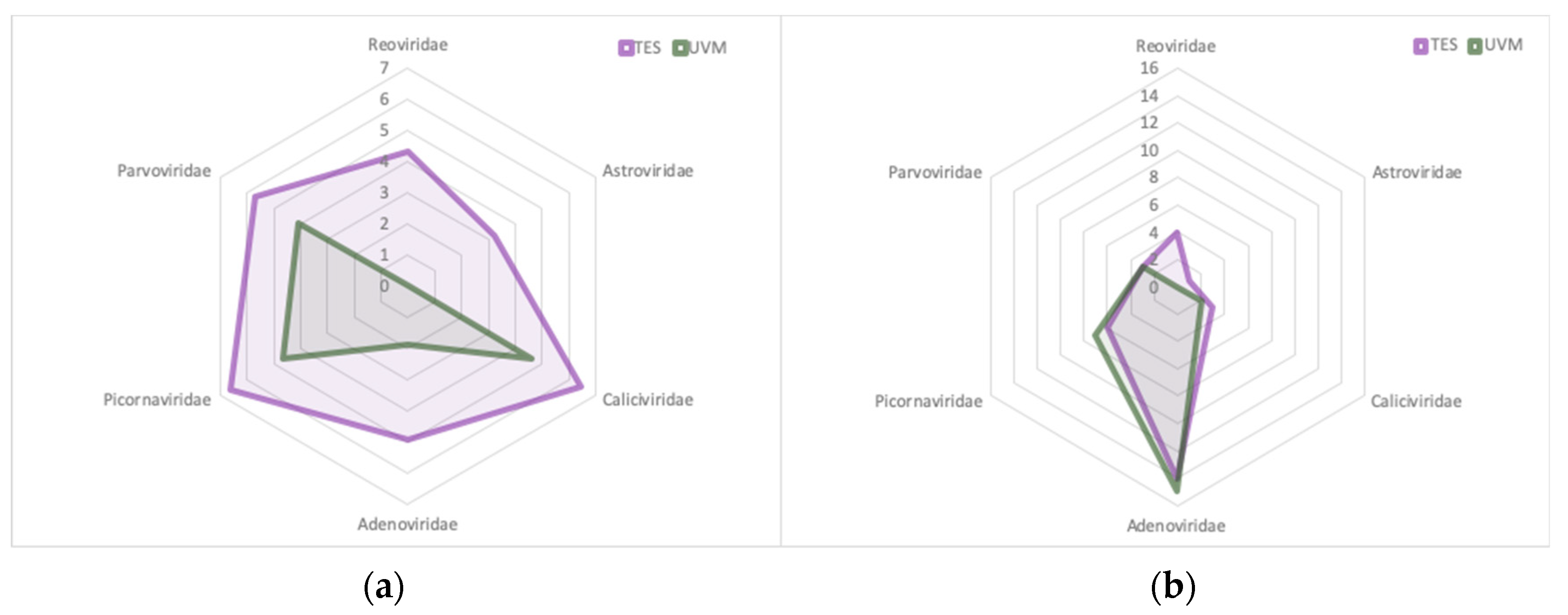
3.1. Metagenomic Identification of Viral Families in Pooled Fecal Samples
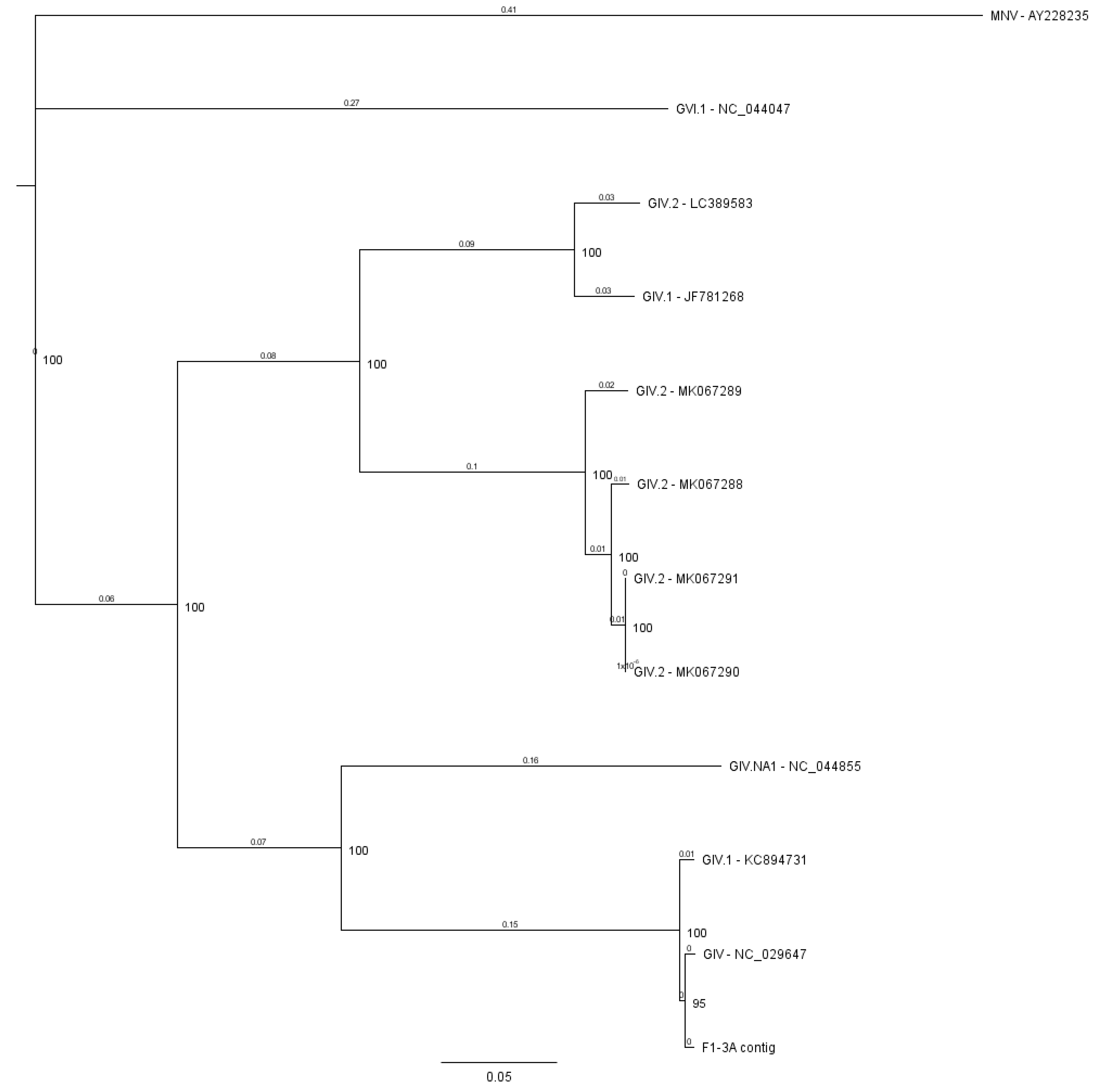
3.2. Caliciviridae
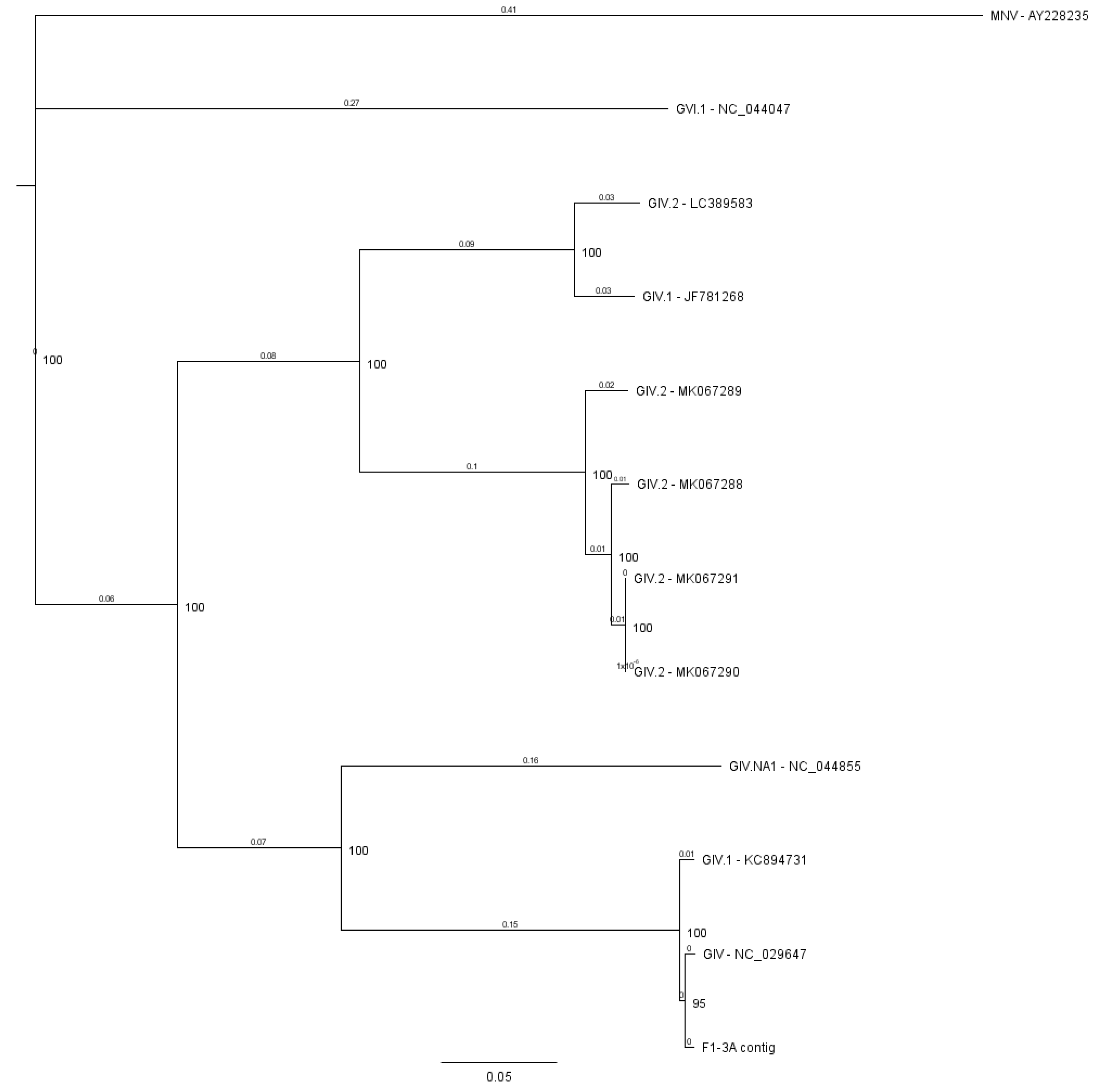
3.2.1. Norovirus
3.2.2. Sapovirus
3.3. Astroviridae
3.4. Adenoviridae
3.5. Reoviridae
3.6. Other Viral Species Identified in Fecal Samples
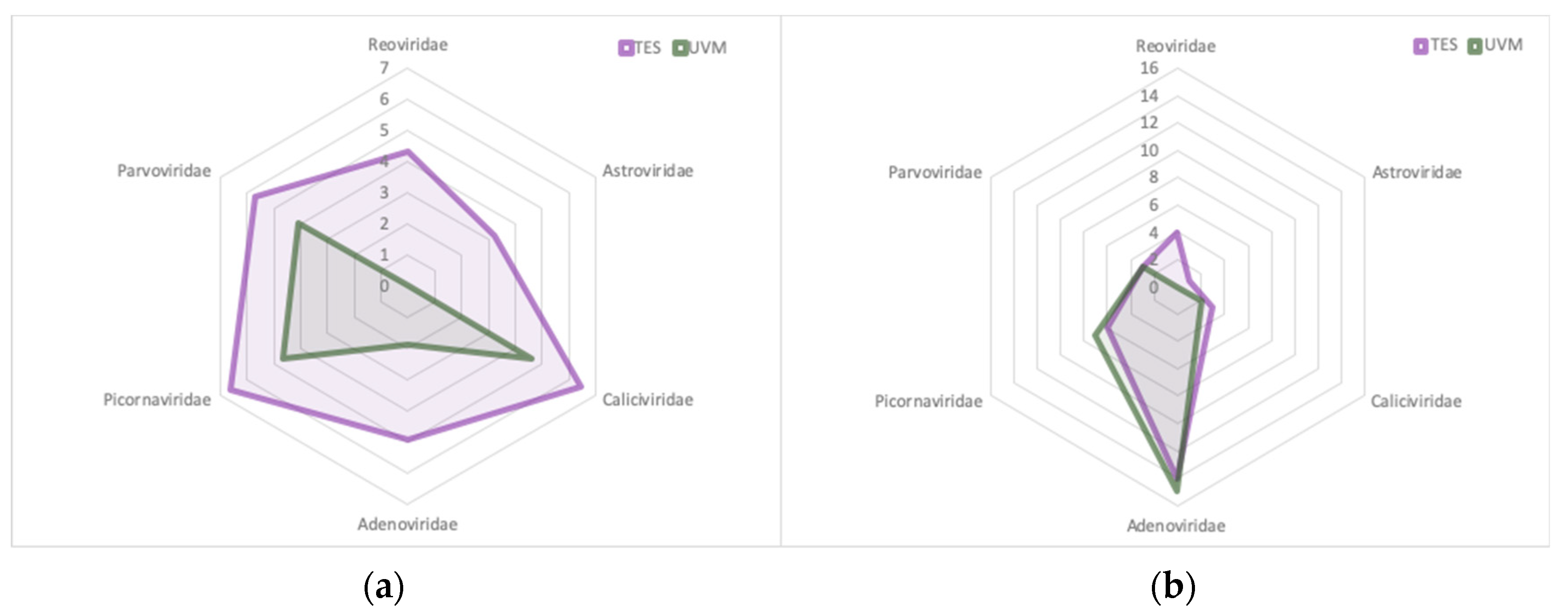
3.7. Target Enrichment Sequencing
4. General Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Material
References
- Troeger, C.; Blacker, B.F.; Khalil, I.A.; Rao, P.C.; Cao, S.; Zimsen, S.R.-M.; Albertson, S.B.; Stanaway, J.D.; Deshpande, A.; Deshpande, A.; et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018, 18, 1211–1228. [Google Scholar] [CrossRef] [Green Version]
- Bányai, K.; Estes, M.K.; Martella, V.; Parashar, U.D. Viral gastroenteritis. Lancet 2018, 392, 175–186. [Google Scholar] [CrossRef]
- Fletcher, S.; Van Hal, S.; Andresen, D.; McLaws, M.L.; Stark, D.; Harkness, J.; Ellis, J. Gastrointestinal pathogen distribution in symptomatic children in Sydney, Australia. J. Epidemiol. Glob. Heal. 2013, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppong, T.B.; Yang, H.; Amponsem-Boateng, C.; Kyere, E.K.D.; Abdulai, T.; Duan, G.; Opolot, G. Enteric pathogens associated with gastroenteritis among children under 5 years in sub-saharan Africa: A systematic review and meta-Analysis. Epidemiol. Infect. 2020, 148, 1–26. [Google Scholar] [CrossRef]
- Tran, A.; Talmud, D.; Lejeune, B.; Jovenin, N.; Renois, F.; Payan, C.; Leveque, N.; Andreoletti, L. Prevalence of rotavirus, adenovirus, norovirus, and astrovirus infections and coinfections among hospitalized children in Northern France. J. Clin. Microbiol. 2010, 48, 1943–1946. [Google Scholar] [CrossRef] [Green Version]
- Chhabra, P.; Payne, D.C.; Szilagyi, P.G.; Edwards, K.M.; Staat, M.A.; Shirley, S.H.; Wikswo, M.; Nix, W.A.; Lu, X.; Parashar, U.D.; et al. Etiology of viral gastroenteritis in children <5 years of age in the United States, 2008–2009. J. Infect. Dis. 2013, 208, 790–800. [Google Scholar] [CrossRef] [Green Version]
- Holtz, L.R.; Finkbeiner, S.R.; Zhao, G.; Kirkwood, C.D.; Girones, R.; Pipas, J.M.; Wang, D. Klassevirus 1, a previously undescribed member of the family Picornaviridae, is globally widespread. Virol. J. 2009, 6, 86. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T.K.; Rasmussen, L.D.; Fonager, J. Taking gastro-surveillance into the 21st century. J. Clin. Virol. 2019, 117, 43–48. [Google Scholar] [CrossRef]
- Kageyama, T.; Kojima, S.; Shinohara, M.; Uchida, K.; Fukushi, S.; Hoshino, F.B.; Takeda, N.; Katayama, K. Broadly reactive and highly sensitive assay for Norwalk-like viruses based on real-time quantitative reverse transcription-PCR. J. Clin. Microbiol. 2003, 41, 1548–1557. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Cassi, X.; Rusiñol, M.; Martínez-Puchol, S. Viral Concentration and Amplification from Human Serum Samples Prior to Application of Next-Generation Sequencing Analysis; Springer: New York, NY, USA, 2018; pp. 173–188. [Google Scholar]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I. Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. mBio 2015, 6, e01491-15. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Puchol, S.; Rusiñol, M.; Fernández-Cassi, X.; Timoneda, N.; Itarte, M.; Andrés, C.; Antón, A.; Abril, J.F.; Girones, R.; Bofill-Mas, S. Characterisation of the sewage virome: Comparison of NGS tools and occurrence of significant pathogens. Sci. Total. Environ. 2020, 713, 136604. [Google Scholar] [CrossRef] [PubMed]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alacantara, L.C.; Vanden Eynden, E.; Vandamme, A.-M.; et al. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, D.A.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2015, 43, D30–D35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Dura, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.W.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393–1406. [Google Scholar] [CrossRef]
- Kroneman, A.; Vennema, H.; Deforche, K.; Avoort, H.; Peñaranda, S.; Oberste, M.S.; Vinjé, J.; Koopmans, M. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 2011, 51, 121–125. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Page, R.D.M. Treeview: An application to display phylogenetic trees on personal computers. Bioinformatics 1996, 12, 157–158. [Google Scholar] [CrossRef] [Green Version]
- Maes, P.; Matthijnssens, J.; Rahman, M.; Van Ranst, M. RotaC: A web-based tool for the complete genome classification of group A rotaviruses. BMC Microbiol. 2009, 9, 238. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, S.; Obermeier, P.E.; Neubauer, K.; Diedrich, S. Recombinant enterovirus A71 subgenogroup C1 strains, Germany, 2015. Emerg. Infect. Dis. 2016, 22, 1843–1846. [Google Scholar] [CrossRef]
- Donato, C.; Vijaykrishna, D. The broad host range and genetic diversity of mammalian and avian astroviruses. Viruses 2017, 9, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loisy, F.; Atmar, R.L.; Guillon, P.; Le Cann, P.; Pommpepuy, M.; Le Guyader, F.S. Real-time RT-PCR for norovirus screening in shellfish. J. Virol. Methods 2005, 123, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Parnaudeau, S.; Grodzki, M.; Okabe, S.; Atmar, R.L.; Le Guyader, F.S. Environmental detection of genogroup I, II, and IV noroviruses by using a generic real-time reverse transcription-PCR assay. Appl. Environ. Microbiol. 2013, 79, 6585–6592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Beek, J.; van der Eijk, A.A.; Fraaij, P.L.A.; Caliskan, K.; Cransberg, K.; Dalinghaus, M.; Hoek, R.A.S.; Metselaar, H.J.; Roodnat, J.; Vennema, H.; et al. Chronic norovirus infection among solid organ recipients in a tertiary care hospital, the Netherlands, 2006–2014. Clin. Microbiol. Infect. 2017, 23, 265.e9–265.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.J.; Eisenbart, V.G.; Etingüe, A.L.; Gould, L.H.; Lopman, B.A.; Parashar, U.D. Epidemiology of foodborne norovirus outbreaks, United States, 2001–2008. Emerg. Infect. Dis. 2012, 18, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Tohma, K.; Lepore, C.J.; Gao, Y.; Ford-Siltz, L.A.; Parra, G.I. Population genomics of gii.4 noroviruses reveal complex diversification and new antigenic sites involved in the emergence of pandemic strains. mBio 2019, 10, e02202-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Beek, J.; de Graaf, M.; Al-Hello, H.; Allen, D.J.; Ambert-Balay, K.; Botteldoorn, N.; Brytting, M.; Buesa, J.; Cabrerizo, M.; Chan, M.; et al. Molecular surveillance of norovirus, 2005–2016: An epidemiological analysis of data collected from the NoroNet network. Lancet Infect. Dis. 2018, 18, 545–553. [Google Scholar] [CrossRef]
- De Graaf, M.; Van Beek, J.; Koopmans, M.P.G. Human norovirus transmission and evolution in a changing world. Nat. Rev. Genet. 2016, 14, 421–433. [Google Scholar] [CrossRef]
- Chen, L.; Xu, D.; Wu, X.; Liu, G.; Ji, L. An increasing prevalence of non-GII.4 norovirus genotypes in acute gastroenteritis outbreaks in Huzhou, China, 2014–2018. Arch. Virol. 2020, 165, 1121–1128. [Google Scholar] [CrossRef]
- Vega, E.; Barclay, L.; Gregoricus, N.; Shirley, S.H.; Lee, D.; Vinjé, J. Genotypic and epidemiologic trends of norovirus outbreaks in the united states, 2009 to 2013. J. Clin. Microbiol. 2013, 52, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lun, J.H.; Hewitt, J.; Yan, G.J.H.; Tuipulotu, D.E.; Rawlinson, W.; White, P. Recombinant GII.P16/GII.4 sydney 2012 was the dominant norovirus identified in Australia and New Zealand in 2017. Viruses 2018, 10, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabrià, A.; Pintó, R.M.; Bosch, A.; Bartolomé, R.; Cornejo, T.; Torner, N.; Martínez, A.; de Simón, M.; Domínguez, A.; Guix, S. Molecular and clinical epidemiology of norovirus outbreaks in Spain during the emergence of GII.4 2012 variant. J. Clin. Virol. 2014, 60, 96–104. [Google Scholar]
- Hoa Tran, T.N.; Trainor, E.; Nakagomi, T.; Cunliffe, N.A.; Nakagomi, O. Molecular epidemiology of noroviruses associated with acute sporadic gastroenteritis in children: Global distribution of genogroups, genotypes and GII.4 variants. J. Clin. Virol. 2013, 56, 269–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mans, J.; Murray, T.Y.; Taylor, M.B. Novel norovirus recombinants detected in South Africa. Virol. J. 2014, 11, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Guyader, F.S.; Atmar, R.L.; Le Pendu, J. Transmission of viruses through shellfish: When specific ligands come into play. Curr. Opin. Virol. 2012, 2, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Cai, H.; Hu, L.; Lei, R.; Pan, Y.; Yan, S.; Wang, Y. Molecular epidemiology of oyster-related human noroviruses and their global genetic diversity and temporal-geographical distribution from 1983 to 2014. Appl. Environ. Microbiol. 2015, 81, 7615–7624. [Google Scholar] [CrossRef] [Green Version]
- Tunyakittaveeward, T.; Rupprom, K.; Pombubpa, K.; Howteerakul, N.; Kittigul, L. Norovirus Monitoring in Oysters Using Two Different Extraction Methods. Food Environ. Virol. 2019, 11, 374–382. [Google Scholar] [CrossRef]
- Kanwar, N.; Hassan, F.; Barclay, L.; Langley, C.; Vinjé, J.; Bryant, P.W.; George, K.; Mosher, L.; Matthews-Greer, J.M.; Rocha, M.A. Evaluation of RIDA ® GENE norovirus GI/GII real time RT-PCR using stool specimens collected from children and adults with acute gastroenteritis. J. Clin. Virol. 2018, 104, 1–4. [Google Scholar] [CrossRef]
- Dunbar, N.L.; Bruggink, L.D.; Marshall, J.A. Evaluation of the RIDAGENE real-time PCR assay for the detection of GI and GII norovirus. Diagn. Microbiol. Infect. Dis. 2014, 79, 317–321. [Google Scholar] [CrossRef]
- Vinjé, J.; Koopmans, M.P.G. Simultaneous detection and genotyping of “Norwalk-like viruses” by oligonucleotide array in a reverse line blot hybridization format. J. Clin. Microbiol. 2000, 38, 2595–2601. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, G.; Pourshaban, M.; Iaconelli, M.; Muscillo, M. Detection of genogroup IV noroviruses in environmental and clinical samples and partial sequencing through rapid amplification of cDNA ends. Arch. Virol. 2008, 153, 2077–2083. [Google Scholar] [CrossRef]
- Ao, Y.Y.; Yu, J.M.; Li, L.L.; Jin, M.; Duan, Z.J. Detection of human norovirus GIV.1 in China: A case report. J. Clin. Virol. 2014, 61, 298–301. [Google Scholar] [CrossRef]
- Muscillo, M.; Fratini, M.; Graffeo, R.; Sanguinetti, M.; Martella, V.; Green, K.Y.; Della Libera, S.; La Rosa, G. GIV Noroviruses in Wastewaters and in Stool Specimens from Hospitalized Patients. Food Environ. Virol. 2013, 5, 194–202. [Google Scholar] [CrossRef]
- Fioretti, J.M.; Fumian, T.M.; Rocha, M.S.; dos Santos, I.D.; Carvalho-Costa, F.A.; de Assis, M.R.; Rodrigues, J.; Leite, J.P.G.; Miagostovich, M.P. Surveillance of Noroviruses in Rio De Janeiro, Brazil: Occurrence of New GIV Genotype in Clinical and Wastewater Samples. Food Environ. Virol. 2017, 10, 1–6. [Google Scholar] [CrossRef]
- Das Neves Costa, L.C.P.; Teixeira, D.M.; Portela, A.C.R.; De Lima, I.C.G.; da Silva Bandeira, R.; Sousa Júnior, E.C.; Siqueira, J.A.M.; Resque, H.R.; da Silva, L.D.; Gabbay, Y.B. Molecular and evolutionary characterization of norovirus gii.17 in the northern region of brazil. BMC Infect. Dis. 2019, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Cai, W.; Gao, J.; Jiang, Y.; Wu, H.; Zhang, L.; Zuo, Y.; Dong, R.; Pang, R.; Zeng, H.; et al. Genome characteristics and molecular evolution of the human sapovirus variant GII.8. Infect. Genet. Evol. 2019, 73, 362–367. [Google Scholar] [CrossRef]
- Varela, M.F.; Rivadulla, E.; Lema, A.; Romalde, J.L. Human sapovirus among outpatients with acute gastroenteritis in Spain: A one-year study. Viruses 2019, 11, 144. [Google Scholar] [CrossRef] [Green Version]
- Mann, P.; Pietsch, C.; Liebert, U.G. Genetic diversity of sapoviruses among inpatients in germany, 2008−2018. Viruses 2019, 11, 726. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Mori, K.; Iritani, N.; Harada, S.; Ueki, Y.; Iizuka, S.; Mise, K.; Murakami, K.; Wakita, T.; Katayama, K. Human sapovirus classification based on complete capsid nucleotide sequences. Arch. Virol. 2012, 157, 349–352. [Google Scholar] [CrossRef]
- Phan, T.G.; Okame, M.; Nguyen, T.A.; Nishio, O.; Okitsu, S.; Ushijima, H. Genetic diversity of sapovirus in fecal specimens from infants and children with acute gastroenteritis in Pakistan. Arch. Virol. 2004, 150, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Usuku, S.; Kumazaki, M. A gastroenteritis outbreak attributed to sapovirus genogroup V in Yokohama, Japan. Jpn. J. Infect. Dis. 2014, 67, 411–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, S.; Sekizuka, T.; Kodaira, A.; Kuroda, M.; Haga, K.; Doan, Y.H.; Takai-Todaka, R.; Katayama, K.; Wakita, T.; Oka, T. Complete genome sequence of a novel GV.2 sapovirus strain, NGY-1, detected from a suspected foodborne gastroenteritis outbreak. Genome Announc. 2015, 3, e01553-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hergens, M.; Nederby Öhd, J.; Alm, E.; Askling, H.H.; Helgesson, S.; Insulander, M.; Lagerqvist, N.; Svenungsson, B.; Tihane, M.; Tolfvenstam, T.; et al. Investigation of a food-borne outbreak of gastroenteritis in a school canteen revealed a variant of sapovirus genogroup V not detected by standard PCR, Sollentuna, Sweden, 2016. Eurosurveillance 2017, 22. [Google Scholar] [CrossRef]
- Jeong, H.S.; Jeong, A.; Cheon, D.S. Epidemiology of astrovirus infection in children. Korean J. Pediatr. 2012, 55, 77–82. [Google Scholar] [CrossRef]
- Cordey, S.; Vu, D.-L.; Schibler, M.; L’Huillier, A.G.; Brito, F.; Docquier, M.; Posfay-Barbe, K.M.; Petty, T.J.; Turin, L.; Zdobnoy, E.M.; et al. Astrovirus MLB2, a New Gastroenteric Virus Associated with Meningitis and Disseminated Infection. Emerg. Infect. Dis. 2016, 22, 846–853. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Li, L.; Victoria, J.; Oderinde, B.; Mason, C.; Pandey, P.; Zaidi, S.Z.; Delwart, E. Multiple novel astrovirus species in human stool. J. Gen. Virol. 2009, 90, 2965–2972. [Google Scholar] [CrossRef]
- Bosch, A.; Pintó, R.M.; Guix, S. Human Astroviruses. Clin. Microbiol. Rev. 2014, 27, 1048–1074. [Google Scholar] [CrossRef] [Green Version]
- Gabbay, Y.B.; Leite, J.P.G.; Oliveira, D.S.; Nakamura, L.S.; Nunes, M.R.T.; Mascarenhas, J.D.A.P.; Heinemann, M.B.; Linhares, A.C. Molecular epidemiology of astrovirus type 1 in Belém, Brazil, as an agent of infantile gastroenteritis, over a period of 18 years (1982–2000): Identification of two possible new lineages. Virus Res. 2007, 129, 166–174. [Google Scholar] [CrossRef]
- Jiang, H.; Holtz, L.R.; Bauer, I.; Franz, C.J.; Zhao, G.; Bodhidatta, L.; Shrestha, S.K.; Kang, G.; Wang, D. Comparison of novel MLB-clade, VA-clade and classic human astroviruses highlights constrained evolution of the classic human astrovirus nonstructural genes. Virology 2013, 436, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.T.; Bauer, I.K.; Antonio, M.; Adeyemi, M.; Saha, D.; Oundo, J.O.; Ochieng, J.B.; Omore, R.; Stine, O.C.; Wang, D.; et al. Prevalence of classic, MLB-clade and VA-clade Astroviruses in Kenya and the Gambia Emerging viruses. Virol. J. 2015, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, D.L.; Cordey, S.; Brito, F.; Kaiser, L. Novel human astroviruses: Novel human diseases? J. Clin. Virol. 2016, 82, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Singh, G.; Lee, J.Y.; Dehghan, S.; Rajaiya, J.; Liu, E.B.; Yousuf, M.A.; Betensky, R.A.; Jones, M.S.; Dyer, D.W. Molecular evolution of human adenoviruses. Scientific Reports 2013, 3, 1812. [Google Scholar] [CrossRef] [Green Version]
- Kaján, G.L.; Kajon, A.E.; Pinto, A.C.; Bartha, D.; Arnberg, N. The complete genome sequence of human adenovirus 84, a highly recombinant new Human mastadenovirus D type with a unique fiber gene. Virus Res. 2017, 242, 79–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hage, E.; Dhingra, A.; Liebert, U.G.; Bergs, S.; Ganzenmueller, T.; Heim, A. Three novel, multiple recombinant types of species of human mastadenovirus D (HAdV-D 73, 74 & 75) isolated from diarrhoeal faeces of immunocompromised patients. J. Gen. Virol. 2017, 98, 3037–3045. [Google Scholar] [PubMed]
- Shimizu, H.; Phan, T.G.; Nishimura, S.; Okitsu, S.; Maneekarn, N.; Ushijima, H. An outbreak of adenovirus serotype 41 infection in infants and children with acute gastroenteritis in Maizuru City, Japan. Infect. Genet. Evol. 2007, 7, 279–284. [Google Scholar] [CrossRef]
- Leruez-Ville, M.; Chardin-Ouachée, M.; Neven, B.; Picard, C.; Le Guinche, I.; Fisher, A.; Rouzioux, C.; Blanche, S. Description of an adenovirus A31 outbreak in a paediatric haematology unit. Bone Marrow Transplant. 2006, 38, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Calcedo, R.; Medina-Jaszek, A.; Keough, M.; Peng, H.; Wilson, J.M. Adenoviruses in lymphocytes of the human gastro-intestinal tract. PLoS ONE 2011, 6, e24859. [Google Scholar] [CrossRef]
- Kosulin, K. Intestinal HAdV Infection: Tissue Specificity, Persistence, and Implications for Antiviral Therapy. Viruses 2019, 11, 804. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, M.F.M.; Fumian, T.M.; de Assis, R.M.S.; Fialho, A.M.; Carvalho-Costa, F.A.; da Silva, J.; Leite, J.P.G. VP7 and VP8* genetic characterization of group A rotavirus genotype G12P[8]: Emergence and spreading in the Eastern Brazilian coast in 2014. J. Med Virol. 2016, 89, 64–70. [Google Scholar] [CrossRef]
- Bányai, K.; László, B.; Duque, J.; Steele, A.D.; Nelson, E.A.S.; Gentsch, J.R.; Parashar, U.D. Systematic review of regional and temporal trends in global rotavirus strain diversity in the pre rotavirus vaccine era: Insights for understanding the impact of rotavirus vaccination programs. Vaccine 2012, 30, A122–A130. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Ciarlet, M.; Heiman, E.; Arijs, I.; Delbeke, T.; McDonald, S.M.; Palombo, E.A.; Iturriza-Gómara, M.; Maes, P.; Patton, J.T. Full Genome-Based Classification of Rotaviruses Reveals a Common Origin between Human Wa-Like and Porcine Rotavirus Strains and Human DS-1-Like and Bovine Rotavirus Strains. J. Virol. 2008, 82, 3204–3219. [Google Scholar] [CrossRef] [Green Version]
- Silva-Sales, M.; Martínez-Puchol, S.; Gonzales-Gustavson, E.; Hundesa, A.; Gironès, R. High prevalence of rotavirus A in raw sewage samples from northeast Spain. Viruses 2020, 12, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutelíková, R.; Dvořáková Heroldová, M.; Holá, V.; Sauer, P.; Prodělalová, J. Human rotavirus a detection: Comparison of enzymatic immunoassay and rapid chromatographic test with two quantitative RT-PCR assays. Epidemiol. Mikrobiol. Imunol. 2018, 63, 110–113. [Google Scholar]
- Khamrin, P.; Tran, D.N.; Chan-it, W.; Thongprachum, A.; Okitsu, S.; Maneekarn, N.; Ushijima, H. Comparison of the rapid methods for screening of group a rotavirus in stool samples. J. Trop. Pediatr. 2011, 57, 375–377. [Google Scholar] [CrossRef]
- Wilcox, A.H.; Delwart, E.; Díaz-Muñoz, S.L. Next-generation sequencing of dsRNA is greatly improved by treatment with the inexpensive denaturing reagent DMSO. Microb. Genom. 2019, 5, e000315. [Google Scholar] [CrossRef]
- Tatte, V.S.; Gopalkrishna, V. Detection of different enteric viruses in children with diarrheal disease: Evidence of the high frequency of mixed infections. Access Microbiol. 2019, 1, e000010. [Google Scholar] [CrossRef]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Arch. Virol. 2016, 161, 2921–3949. [Google Scholar] [CrossRef] [Green Version]
- Nikonov, O.S.; Chernykh, E.S.; Garber, M.B.; Nikonova, E.Y. Enteroviruses: Classification, diseases they cause, and approaches to development of antiviral drugs. Biochemistry 2017, 82, 1615–1631. [Google Scholar] [CrossRef]
- Arakawa, M.; Okamoto-Nakagawa, R.; Toda, S.; Tsukagoshi, H.; Kobayashi, M.; Ryo, A.; Mizuta, K.; Hasegawa, S.; Hirano, R.; Wakiguchi, H.; et al. Molecular epidemiological study of human rhinovirus species A, B and C from patients with acute respiratory illnesses in Japan. J. Med Microbiol. 2012, 61, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.T.; Kuypers, J.; Chu, H.Y.; Foote, S.; Hashikawa, A.; Fairchok, M.P.; Englund, J.A. Heterotypic infection and spread of rhinovirus a, b, and c among childcare attendees. J. Infect. Dis. 2018, 218, 848–855. [Google Scholar] [CrossRef]
- Daleno, C.; Piralla, A.; Scala, A.; Senatore, L.; Principi, N.; Esposito, S. Phylogenetic analysis of human rhinovirus isolates collected from otherwise healthy children with community-acquired pneumonia during five successive years. PLoS ONE 2013, 8, e80614. [Google Scholar] [CrossRef] [Green Version]
- Olijve, L.; Jennings, L.; Walls, T. Human parechovirus: An increasingly recognized cause of sepsis-like illness in young infants. Clin. Microbiol. Rev. 2017, 31, e00047-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.S.; Lukashov, V.V.; Ganac, R.D.; Schnurr, D.P. Discovery of a novel human picornavirus in a stool sample from a pediatric patient presenting with fever of unknown origin. J. Clin. Microbiol. 2007, 45, 2144–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.L.; Liu, N.; Yu, J.M.; Ao, Y.Y.; Li, S.; Stine, O.C.; Duan, Z.J. Analysis of Aichi virus and Saffold virus association with pediatric acute gastroenteritis. J. Clin. Virol. 2017, 87, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Vandesande, H.; Edman, K.; Rondahl, E.; Falkeborn, T.; Serrander, L.; Lindberg, A.M. Saffold virus infection in elderly people with acute gastroenteritis in Sweden. J. Med Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bonanno Ferraro, G.; Mancini, P.; Veneri, C.; Iaconelli, M.; Suffredini, E.; Brandtner, D.; La Rosa, G. Evidence of Saffold virus circulation in Italy provided through environmental surveillance. Lett. Appl. Microbiol. 2019, 70, 102–108. [Google Scholar] [CrossRef]
- Fernandez-Cassi, X.; Timoneda, N.; Martínez-Puchol, S.; Rusiñol, M.; Rodriguez-Manzano, J.; Figuerola, N.; Bofill-Mas, S.; Abril, J.F.; Girones, R. Metagenomics for the study of viruses in urban sewage as a tool for public health surveillance. Sci. Total. Environ. 2018, 618, 870–880. [Google Scholar] [CrossRef]
- Reuter, G.; Boros, Á.; Pankovics, P. Kobuviruses—A comprehensive review. Rev. Med Virol. 2011, 21, 32–41. [Google Scholar] [CrossRef]
- Ambert-Balay, K.; Lorrot, M.; Bon, F.; Giraudon, H.; Kaplon, J.; Wolfer, M.; Lebon, P.; Gendrel, D.; Pothier, P. Prevalence and genetic diversity of Aichi virus strains in stool samples from community and hospitalized patients. J. Clin. Microbiol. 2008, 46, 1252–1258. [Google Scholar] [CrossRef] [Green Version]
- Rivadulla, E.; Romalde, J.L. A Comprehensive Review on Human Aichi Virus. Virol. Sin. 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Khamrin, P.; Maneekarn, N.; Okitsu, S.; Ushijima, H. Epidemiology of human and animal kobuviruses. VirusDisease 2014, 25, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, S.R.; Wang, D. Extensive conservation of prokaryotic ribosomal binding sites in known and novel picobirnaviruses. Virology 2018, 516, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Leitão, G.A.A.; Olivares, A.I.O.; Pimenta, Y.C.; Delgado, I.F.; Miagostovich, M.P.; Leite, J.P.G.; Moraes, M.T.B. Human Bocavirus genotypes 1 and 2 detected in younger Amazonian children with acute gastroenteritis or respiratory infections, respectively. Int. J. Infect. Dis. 2020, 95, 32–37. [Google Scholar] [CrossRef]
- Haloschan, M.; Bettesch, R.; Görzer, I.; Weseslindtner, L.; Kundi, M.; Puchhammer-Stöckl, E. TTV DNA plasma load and its association with age, gender, and HCMV IgG serostatus in healthy adults. Age 2014, 36, 9716. [Google Scholar] [CrossRef] [Green Version]
- Focosi, D.; Antonelli, G.; Pistello, M.; Maggi, F. Torquetenovirus: The human virome from bench to bedside. Clin. Microbiol. Infect. 2016, 22, 589–593. [Google Scholar] [CrossRef] [Green Version]
- Wildenbeest, J.G.; Benschop, K.S.M.; Bouma-De Jongh, S.; Wolthers, K.C.; Pajkrt, D. Prolonged Shedding of Human Parechovirus in Feces of Young Children after Symptomatic Infection. Pediatr. Infect. Dis. J. 2016, 35, 580–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netshikweta, R.; Chidamba, L.; Nadan, S.; Taylor, M.B.; Page, N.A. Molecular epidemiology of human bocavirus infection in hospitalized children with acute gastroenteritis in South Africa, 2009–2015. J. Med Virol. 2019, 92, 1124–1132. [Google Scholar] [CrossRef]
- De, R.; Liu, L.; Qian, Y.; Zhu, R.; Deng, J.; Wang, F.; Sun, Y.; Dong, H.; Jia, L.; Zhao, L. Risk of acute gastroenteritis associated with human bocavirus infection in children: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0184833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paskey, A.C.; Frey, K.G.; Schroth, G.; Gross, S.; Hamilton, T.; Bishop-Lilly, K.A. Enrichment post-library preparation enhances the sensitivity of high-throughput sequencing-based detection and characterization of viruses from complex samples. BMC Genom. 2019, 20, 155. [Google Scholar] [CrossRef]
- Luchs, A.; do Carmo Sampaio Tavares Timenetsky, M. Group A rotavirus gastroenteritis: Post-vaccine era, genotypes and zoonotic transmission. Einstein 2016, 14, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus–host interactions resolved from publicly available microbial genomes. eLife 2015, 4, e08490. [Google Scholar] [CrossRef] [PubMed]
- Clooney, A.G.; Sutton, T.D.S.; Shkoporov, A.N.; Holohan, R.K.; Daly, K.M.; O’Regan, O.; Ryan, F.J.; Draper, L.A.; Plevy, S.E.; Ross, R.P.; et al. Whole-Virome Analysis Sheds Light on Viral Dark Matter in Inflammatory Bowel Disease. Cell Host Microbe 2019, 26, 764–778.e5. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Bacterial Parameters | Viral Parameters | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pool | Age | N | Salmonella sp. | Campylobacter sp. | Shigella sp. | Yersinia sp. | Aeromonas sp. | Verotoxigenic E. coli (VTEC) | Vibrio sp. | Rotavirus | Adenovirus | Astrovirus | Norovirus GGI | Norovirus GGII |
| F < 1 | <1 | 10 | 0/9 | 0/9 | 0/9 | 0/9 | 0/0 | 0/0 | 0/0 | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 |
| S < 1A | 9 | 0/9 | 0/9 | 0/9 | 0/9 | 0/7 | 0/0 | 0/8 | 0/9 | 0/9 | 0/9 | 0/0 | 0/0 | |
| S < 1B | 9 | 0/9 | 0/9 | 0/9 | 0/9 | 0/8 | 0/0 | 0/8 | 0/9 | 0/9 | 0/9 | 0/1 | 0/1 | |
| F1-3A | 1–3 | 23 | 0/23 | 0/23 | 0/23 | 0/23 | 0/0 | 1/1 | 0/1 | 0/23 | 0/23 | 0/23 | 0/23 | 0/23 |
| S1-3A | 14 | 0/13 | 0/13 | 0/13 | 0/13 | 0/11 | 0/1 | 0/11 | 0/13 | 0/13 | 0/13 | 0/1 | 0/1 | |
| S1-3B | 14 | 0/14 | 0/14 | 0/14 | 0/14 | 0/7 | 0/0 | 0/8 | 0/14 | 0/14 | 0/14 | 0/1 | 0/1 | |
| S1-3C | 13 | 0/14 | 0/14 | 0/14 | 0/14 | 0/8 | 0/0 | 0/11 | 0/14 | 0/14 | 0/14 | 0/0 | 0/0 | |
| F3-10 | 3–10 | 13 | 0/13 | 0/13 | 0/13 | 0/13 | 0/0 | 0/0 | 0/0 | 0/12 | 0/12 | 0/13 | 0/13 | 0/13 |
| S3-10 | 10 | 0/9 | 0/9 | 0/9 | 0/9 | 0/9 | 0/0 | 0/9 | 0/1 | 0/10 | 0/10 | 0/0 | 0/0 | |
| F > 10 | >10 | 9 | 0/9 | 0/9 | 0/9 | 0/9 | 0/0 | 0/1 | 0/1 | 0/8 | 0/8 | 0/9 | 0/9 | 0/9 |
| Pool | Contig Length | Classification | Polymerase Genotype | Capsid Genotype | BLAST Score |
|---|---|---|---|---|---|
| F < 1 | 7521 | Sapovirus GV | NT | GV.1 | 98.03 |
| S < 1A | 7440 | Norovirus GII | GII.P21 (GII.Pb) | GII.3 | 80.95 |
| F1-3A | 7187 | Norovirus GIV | NT | NT | 77.26 |
| 4827 | Sapovirus GI | NT | GI.2 | 73.61 | |
| S1-3A | 5978 | Norovirus GII | GII.P31 (GII.Pe) | GII.2 | 85.10 |
| 1107 | Norovirus GII | NT | NT | 88.89 | |
| 7292 | Sapovirus GII | NT | GII.1b | 74.37 | |
| 550 | Sapovirus GI | NT | NT | 74.27 | |
| 376 | Sapovirus GI | NT | NT | 74.86 | |
| 698 | Sapovirus GI | NT | NT | 76.72 | |
| 2109 | Sapovirus GI | NT | NT | 72.10 | |
| 859 | Sapovirus GI | NT | GI.3 | 78.06 | |
| 1321 | Sapovirus GI | NT | GI.3 | 71.14 | |
| S1-3B | 6808 | Norovirus GII | GII.P17 | GII.17 | 75.56 |
| F3-10 | 7384 | Norovirus GII | GII.P4 New_Orleans_2009 | GII.4 New_Orleans_2009 | 84.54 |
| 7521 | Sapovirus GV | NT | GV.1 | 98.08 | |
| S3-10 | 3533 | Norovirus GI | NT | NT | |
| 2065 | Norovirus GI | NT | GI.2 | 69.38 | |
| 798 | Norovirus GI | GI.P2 | GI.2 | 77.26 | |
| 1551 | Sapovirus GI | NT | NT | 75.02 | |
| 1059 | Sapovirus GI | NT | NT | 72.61 | |
| 345 | Sapovirus GI | NT | GI.3 | 66.47 | |
| 349 | Sapovirus GI | NT | GI.3 | 73.99 | |
| F > 10 | 924 | Norovirus GIV | NT | NT | 98.70 |
| 576 | Norovirus GIV | NT | NT | 98.44 | |
| 1249 | Norovirus GIV | NT | NT | 99.25 | |
| 375 | Norovirus GIV | NT | NT | 98.67 |
| Pool | Contig Length | Classification | ORF2 Genotype | % ORF2 Identity | % ORF2 Coverage | % Genome Identity | % Genome Coverage |
|---|---|---|---|---|---|---|---|
| F < 1 | 6569 | Mamastrovirus 6 | MLB-3 | 97.8 | 100 | 98.1 | 99.8 |
| S1-3A | 6642 | Mamastrovirus 1 | HAstV-2 | 92.7 | 100 | 85.7 | 97.6 |
| F3-10 | 6652 | Mamastrovirus 1 | HAstV-1 | 95.4 | 100 | 94.1 | 97.6 |
| F > 10 | 6651 | Mamastrovirus 1 | HAstV-1 | 95.1 | 100 | 94.0 | 97.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Cassi, X.; Martínez-Puchol, S.; Silva-Sales, M.; Cornejo, T.; Bartolome, R.; Bofill-Mas, S.; Girones, R. Unveiling Viruses Associated with Gastroenteritis Using a Metagenomics Approach. Viruses 2020, 12, 1432. https://doi.org/10.3390/v12121432
Fernandez-Cassi X, Martínez-Puchol S, Silva-Sales M, Cornejo T, Bartolome R, Bofill-Mas S, Girones R. Unveiling Viruses Associated with Gastroenteritis Using a Metagenomics Approach. Viruses. 2020; 12(12):1432. https://doi.org/10.3390/v12121432
Chicago/Turabian StyleFernandez-Cassi, Xavier, Sandra Martínez-Puchol, Marcelle Silva-Sales, Thais Cornejo, Rosa Bartolome, Silvia Bofill-Mas, and Rosina Girones. 2020. "Unveiling Viruses Associated with Gastroenteritis Using a Metagenomics Approach" Viruses 12, no. 12: 1432. https://doi.org/10.3390/v12121432
APA StyleFernandez-Cassi, X., Martínez-Puchol, S., Silva-Sales, M., Cornejo, T., Bartolome, R., Bofill-Mas, S., & Girones, R. (2020). Unveiling Viruses Associated with Gastroenteritis Using a Metagenomics Approach. Viruses, 12(12), 1432. https://doi.org/10.3390/v12121432