
Phylogenomic Analysis of Global Isolates of Canid Alphaherpesvirus 1

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Sequencing
2.3. Genome Assembly
2.4. Viral Genome Alignments
2.5. Variant Analysis
2.6. Phylogenetic and Recombination Analysis
2.7. Sequence Accession Numbers
3. Results
3.1. Sequencing and Genome Assembly
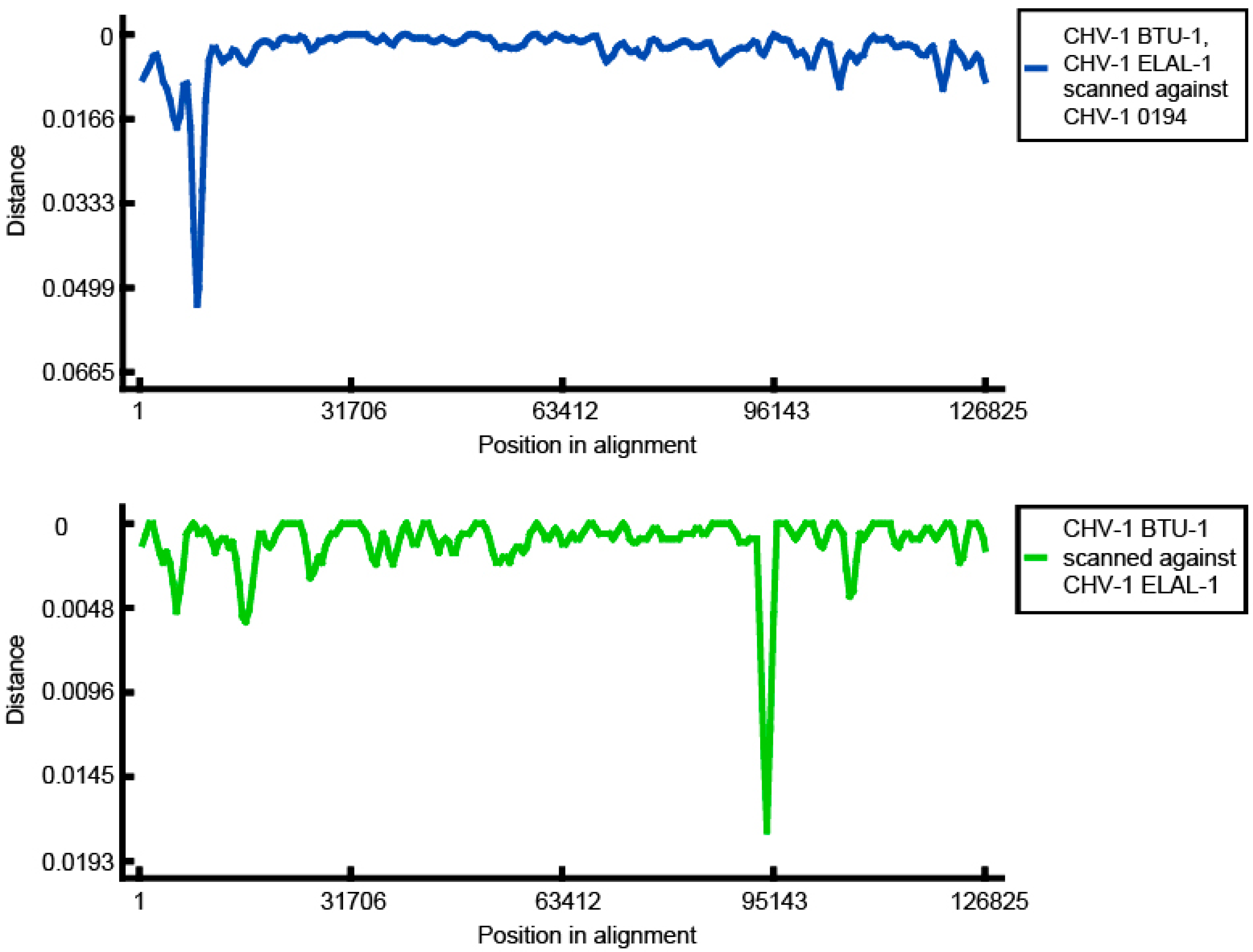
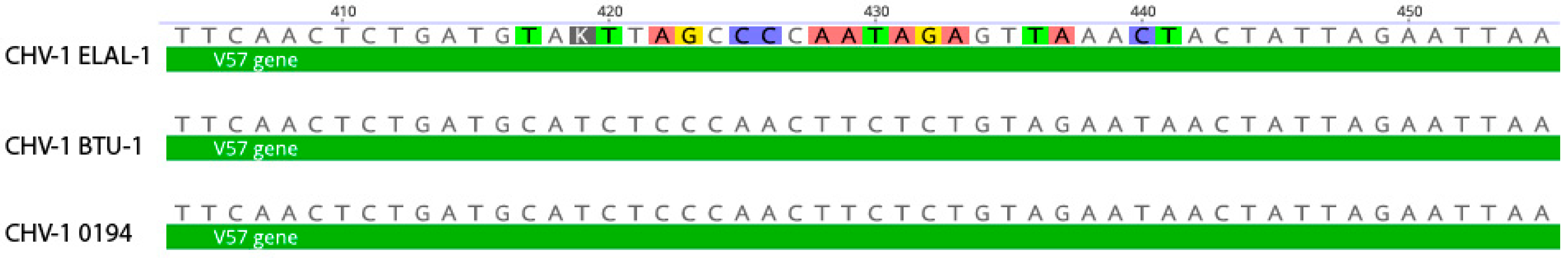
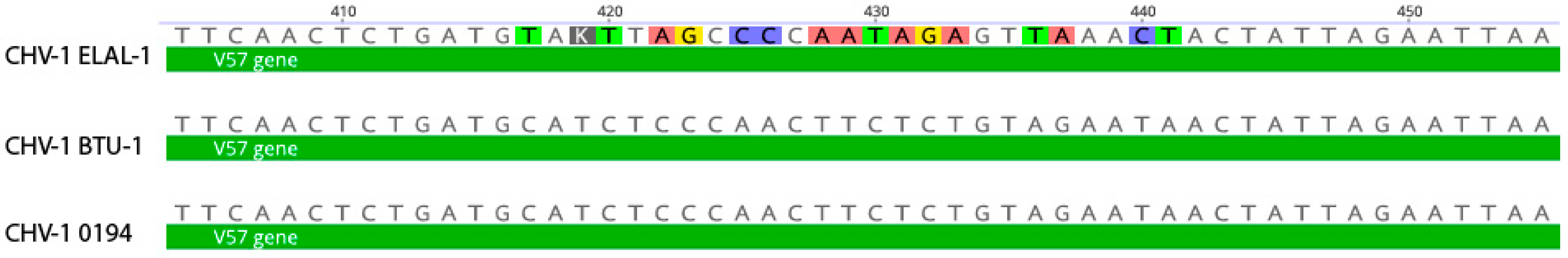
3.2. Variant Analysis
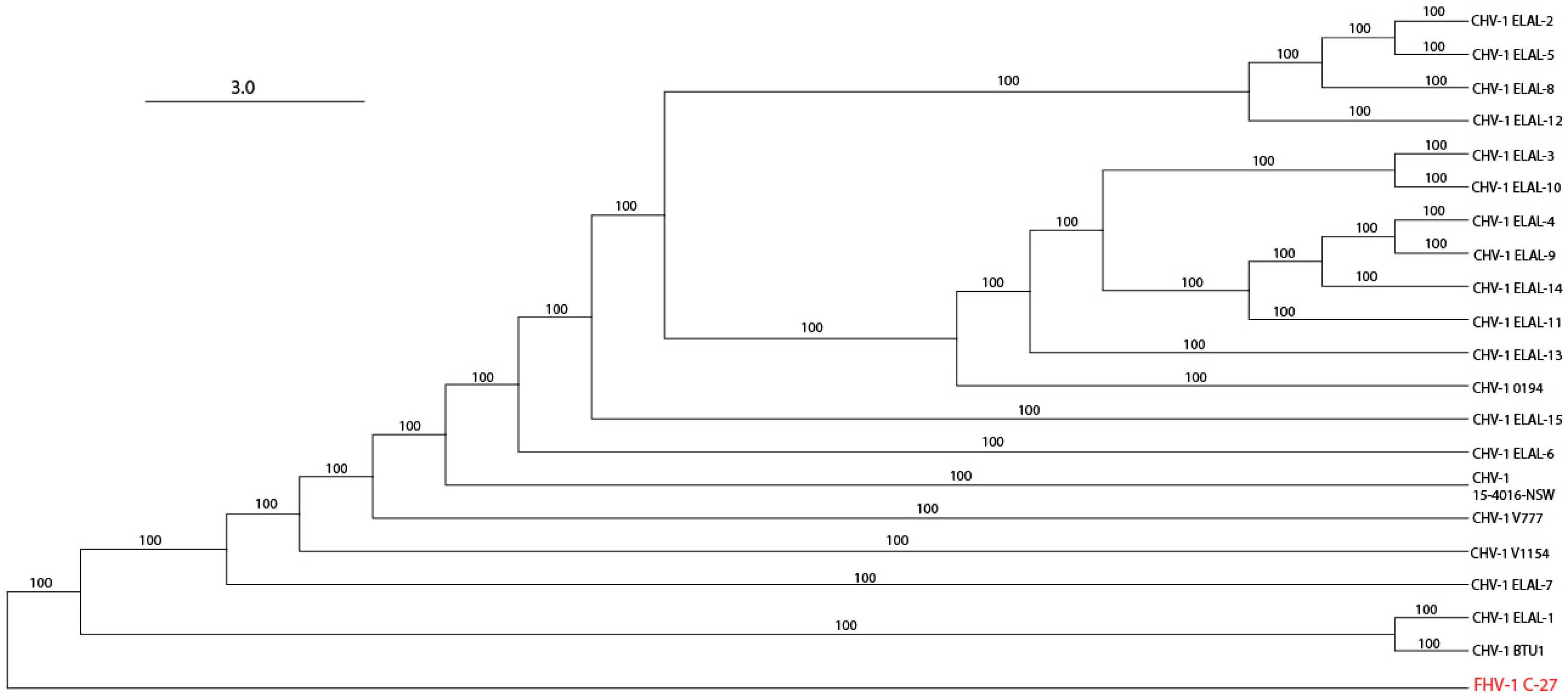
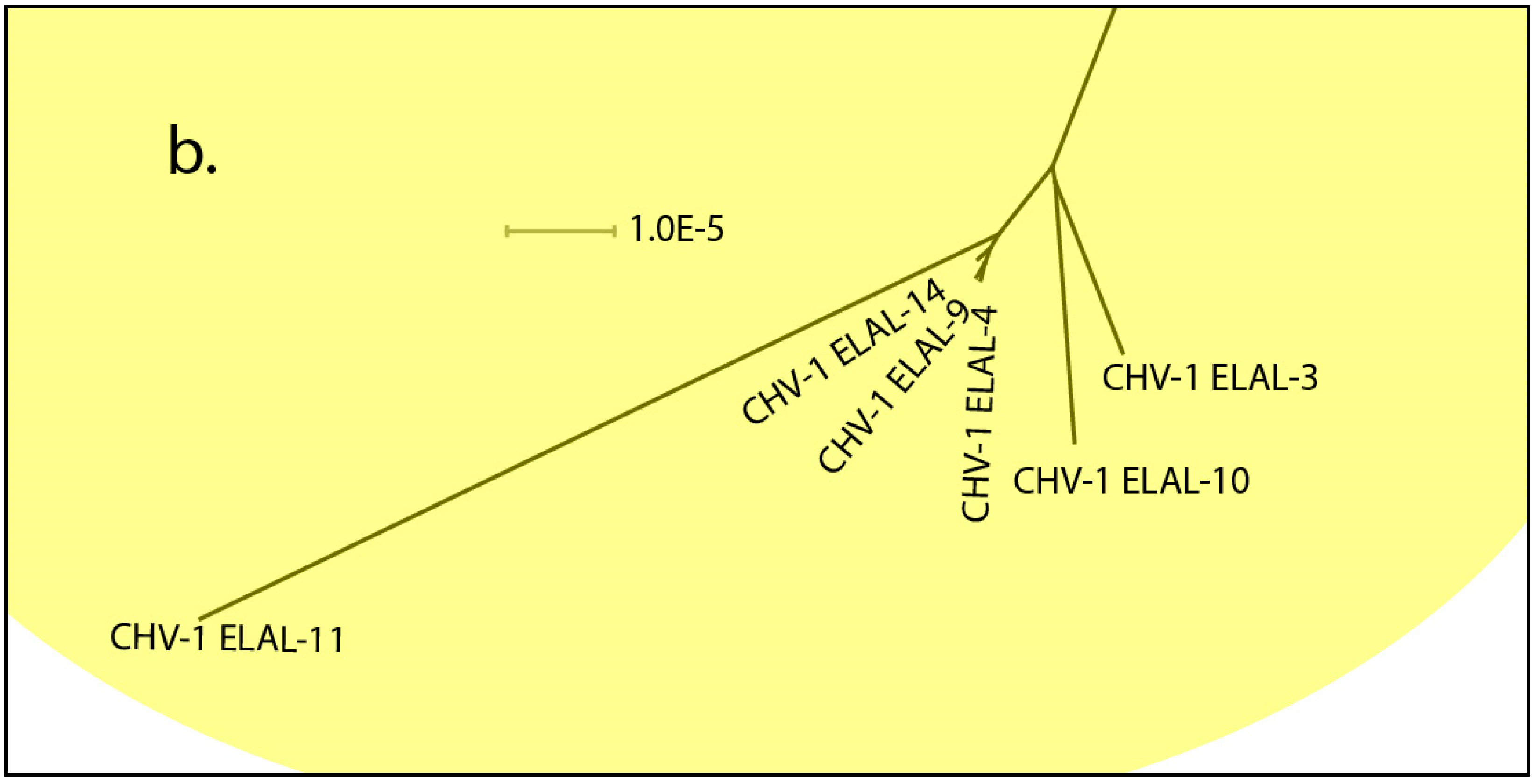
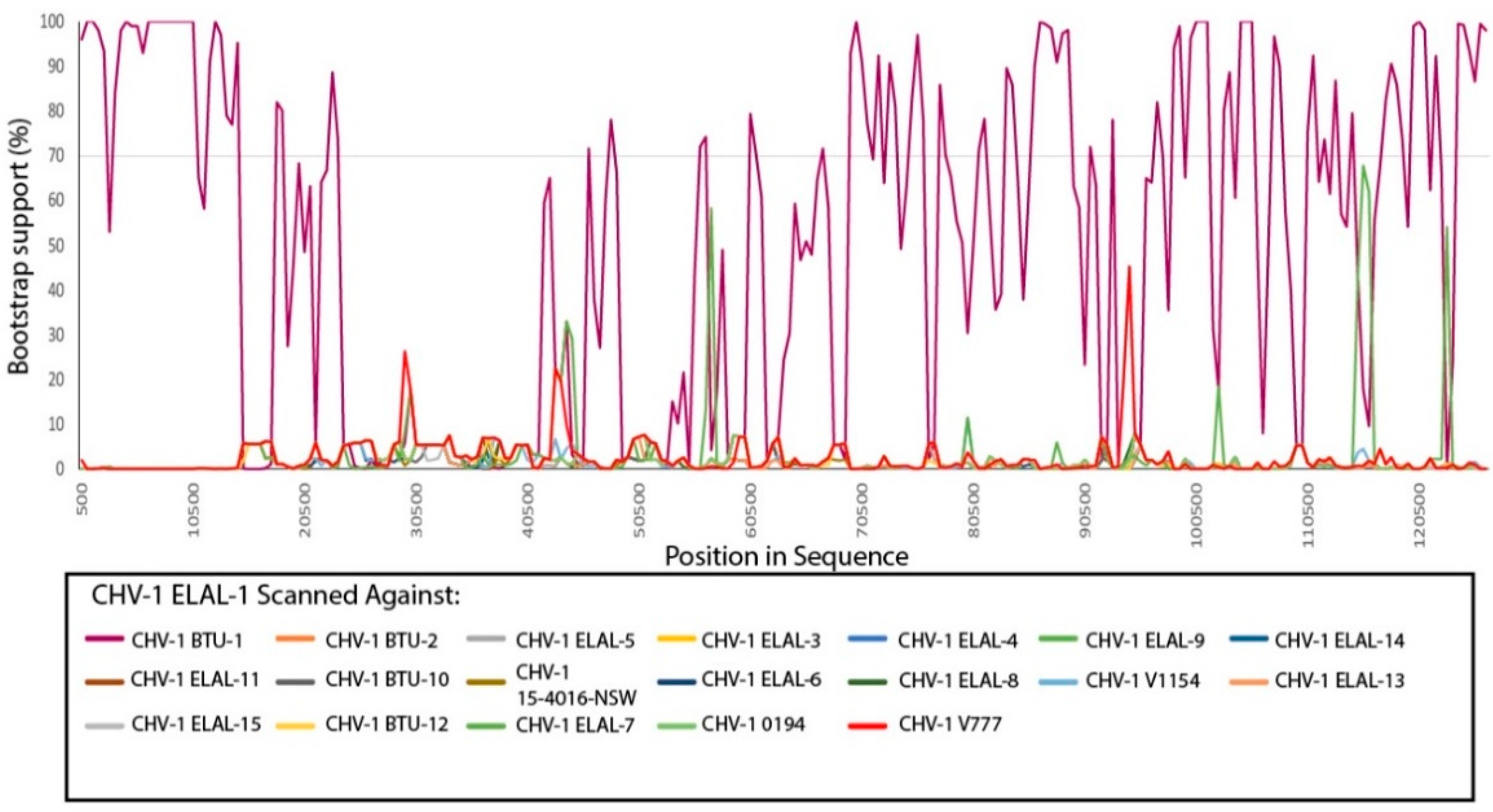
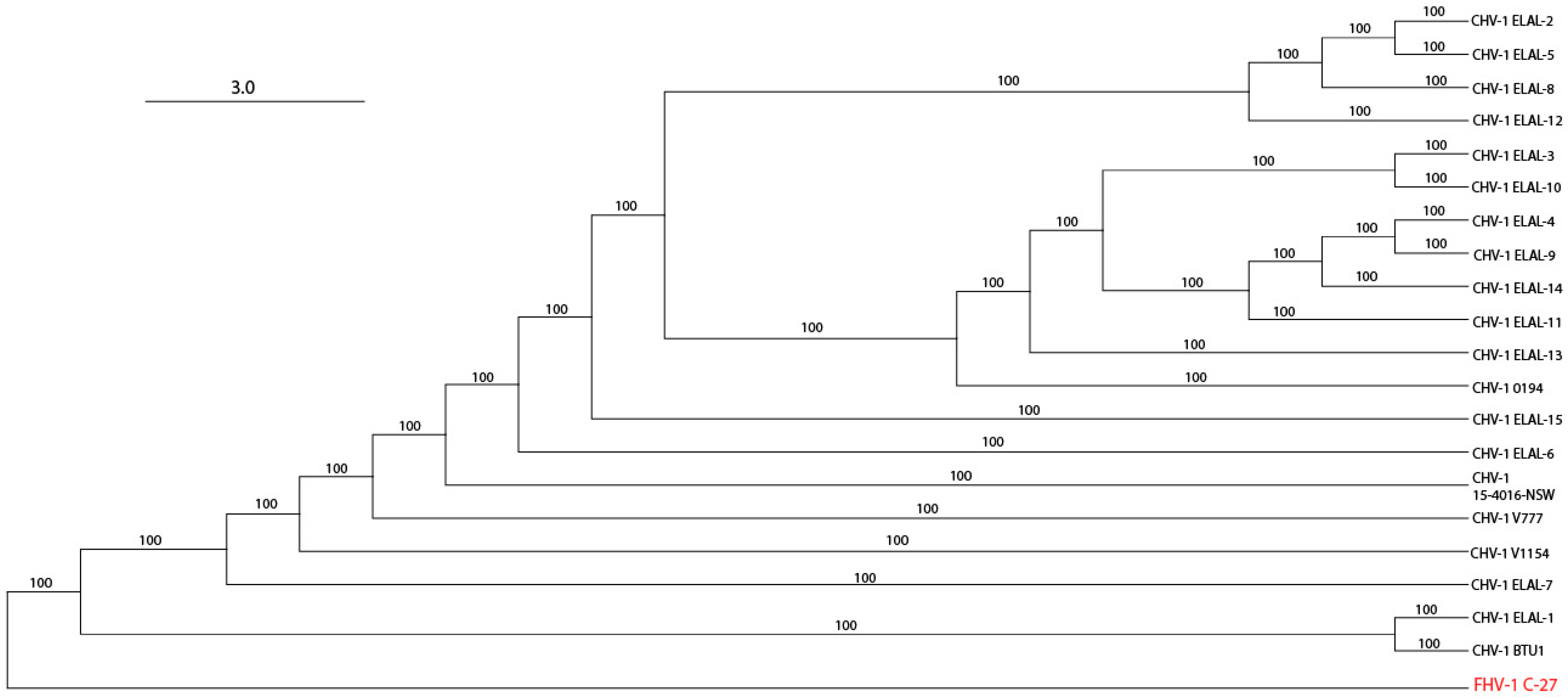
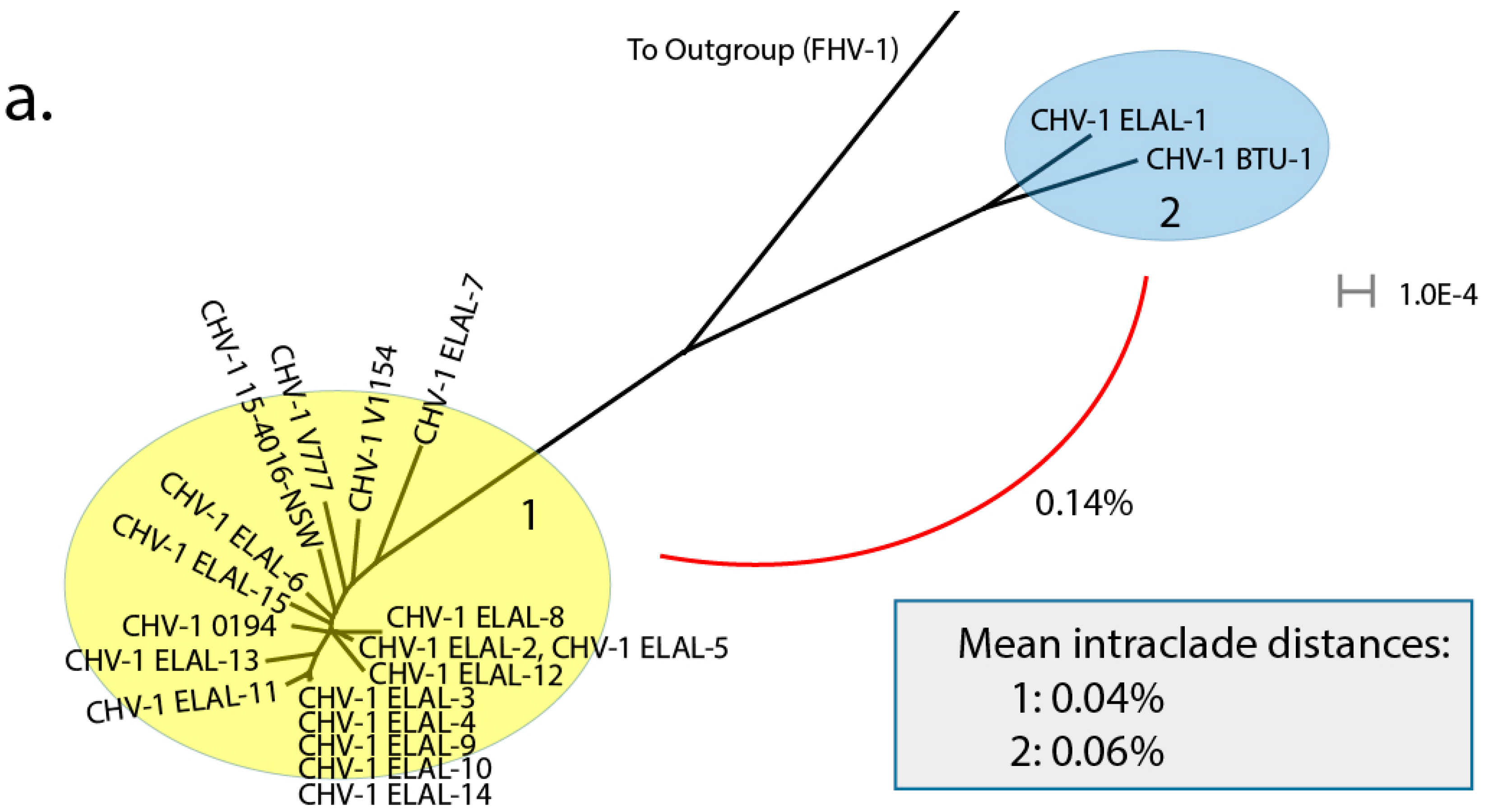
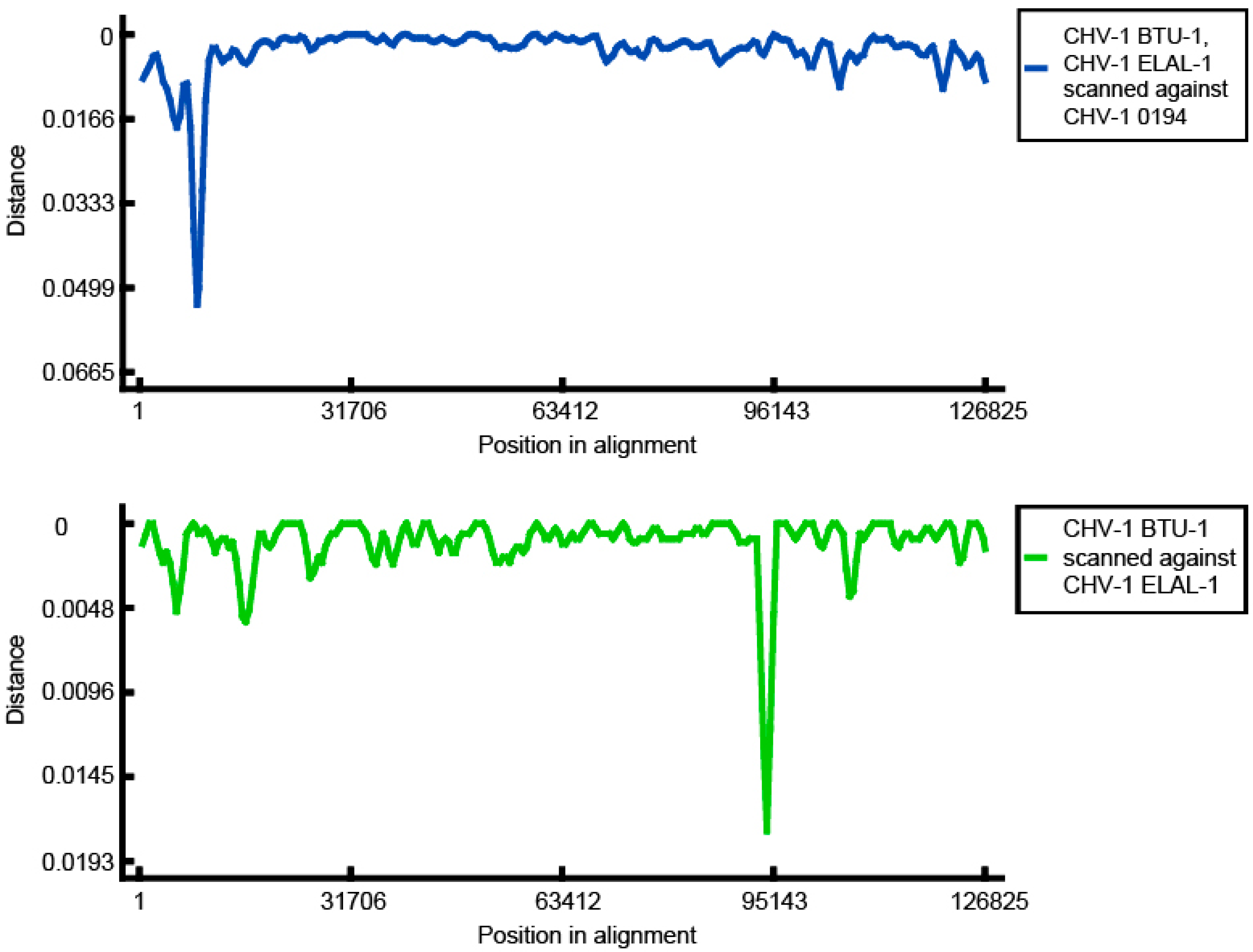
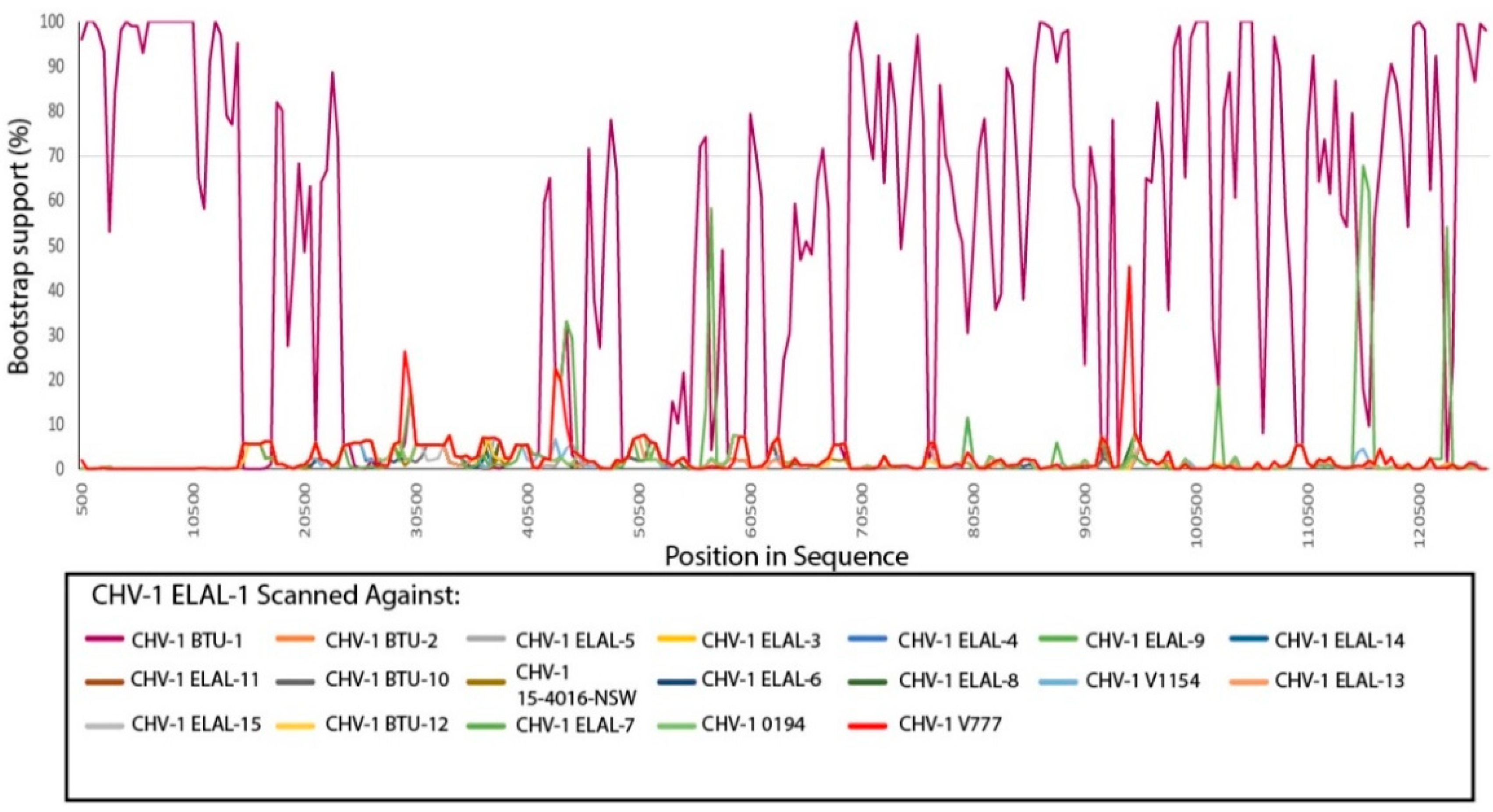
3.3. Phylogenetic and Recombination Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Engels, M.; Mayrbibrack, B.; Ruckstuhl, B.; Metzler, A.; Wyler, R. The Sero-Epizootiology of Canine Herpes-Virus Infections in Switzerland and Preliminary Studies with a Vaccine. Zent. Vet. Reihe B (Ger. FR) 1980, 27, 257–267. [Google Scholar]
- Reading, M.J.; Field, H.J. Detection of high levels of canine herpes virus-1 neutralising antibody in kennel dogs using a novel serum neutralisation test. Res. Vet. Sci. 1999, 66, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Van Gucht, S.; Nauwynck, H.; Pensaert, M. Prevalence of canine herpesvirus in kennels and the possible association with fertility problems and neonatal death. Vlaams Diergeneeskd. Tijdschr. 2001, 70, 204–211. [Google Scholar]
- Carmichael, L.E.; Fabricant, J.; Squire, R.A. A Fatal Septicemic Disease of Infant Puppies Caused by Cytopathogenic Organisms with Characteristics of Mycoplasma. Proc. Soc. Exp. Biol. Med. 1964, 117, 826–833. [Google Scholar] [CrossRef]
- Ledbetter, E.C. Canine herpesvirus-1 ocular diseases of mature dogs. N. Z. Vet. J. 2013, 61, 193–201. [Google Scholar] [CrossRef]
- Karpas, A.; Garcia, F.G.; Calvo, F.; Cross, R.E. Experimental Production of Canine Tracheobronchitis (Kennel Cough) with Canine Herpesvirus Isolated from Naturally Infected Dogs. Am. J. Vet. Res. 1968, 29, 1251–1257. [Google Scholar] [PubMed]
- Wright, N.G.; Cornwell, H.J. Susceptibility of 6-Week Old Puppies to Canine Herpes Virus. J. Small Anim. Pract. 1970, 10, 669–674. [Google Scholar] [CrossRef]
- Poste, G.; King, N. Isolation of a Herpesvirus from Canine Genital Tract—Association with Infertility, Abortion and Stillbirths. Vet. Rec. 1971, 88, 229. [Google Scholar] [CrossRef]
- Hashimoto, A.; Hirai, K.; Fukushi, H.; Fujimoto, Y. The Vaginal Lesions of a Bitch with a History of Canine Herpesvirus-Infection. Jpn. J. Vet. Sci. 1983, 45, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Burr, P.D.; Campbell, M.E.M.; Nicolson, L.; Onions, D.E. Detection of canine herpesvirus 1 in a wide range of tissues using the polymerase chain reaction. Vet. Microbiol. 1996, 53, 227–237. [Google Scholar] [CrossRef]
- Schulze, C.; Baumgartner, W. Nested polymerase chain reaction and in situ hybridization for diagnosis of canine herpesvirus infection in puppies. Vet. Pathol. 1998, 35, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacheretz, A.; Cognard, S. Epidemiology and serological diagnosis of canine herpesvirus infection. Rev. Med. Vet. 1998, 149, 853–856. [Google Scholar]
- Reading, M.J.; Field, H.J. A serological study of canine herpes virus-1 infection in the English dog population. Arch. Virol. 1998, 143, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Rijsewijk, F.A.M.; Luiten, E.J.; Daus, F.J.; van der Heijden, R.W.; van Oirschot, J.T. Prevalence of antibodies against canine herpesvirus 1 in dogs in The Netherlands in 1997–1998. Vet. Microbiol. 1999, 65, 1–7. [Google Scholar] [CrossRef]
- Ledbetter, E.C.; Nicklin, A.M.; Spertus, C.B.; Pennington, M.R.; Van de Walle, G.R.; Mohammed, H.O. Evaluation of topical ophthalmic ganciclovir gel for the treatment of dogs with experimentally induced ocular canine herpesvirus-1 infection. Am. J. Vet. Res. 2018, 79, 762–769. [Google Scholar] [CrossRef]
- Poulet, H.; Guigal, P.M.; Soulier, M.; Leroy, V.; Fayet, G.; Minke, J.; Merial, G.C. Protection of puppies against canine herpesvirus by vaccination of the dams. Vet. Rec. 2001, 148, 691–695. [Google Scholar] [CrossRef]
- Ledbetter, E.C.; Kim, K.; Dubovi, E.J.; Mohammed, H.O.; Felippe, M.J.B. Clinical and immunological assessment of therapeutic immunization with a subunit vaccine for recurrent ocular canine herpesvirus-1 infection in dogs. Vet. Microbiol. 2016, 197, 102–110. [Google Scholar] [CrossRef]
- Papageorgiou, K.V.; Suárez, N.M.; Wilkie, G.S.; McDonald, M.; Graham, E.M.; Davison, A.J. Genome Sequence of Canine Herpesvirus. PLoS ONE 2016, 11, e0156015. [Google Scholar] [CrossRef] [Green Version]
- Sarker, S.; Das, S.; Helbig, K.; Peters, A.; Raidal, S.R. Genome sequence of an Australian strain of canid alphaherpesvirus 1. Aust. Vet. J. 2018, 96, 24–27. [Google Scholar] [CrossRef]
- Kolb, A.W.; Lewin, A.C.; Moeller Trane, R.; McLellan, G.J.; Brandt, C.R. Phylogenetic and recombination analysis of the herpesvirus genus varicellovirus. BMC Genom. 2017, 18, 1–17. [Google Scholar] [CrossRef]
- Lewin, A.C.; Kolb, A.W.; McLellan, G.J.; Bentley, E.; Bernard, K.A.; Newbury, S.P.; Brandt, C.R. Genomic, Recombinational and Phylogenetic Characterization of Global Feline Herpesvirus 1 Isolates. Virology 2018, 518, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Ledbetter, E.C.; Riis, R.C.; Kern, T.J.; Haley, N.J.; Schatzberg, S.J. Corneal ulceration associated with naturally occurring canine herpesvirus-1 infection in two adult dogs. J. Am. Vet. Med. Assoc. 2006, 229, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Nei, M. Limitations of the evolutionary parsimony method of phylogenetic analysis. Mol. Biol. Evol. 1990, 7, 82–102. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Thomasy, S.M.; Maggs, D.J. A review of antiviral drugs and other compounds with activity against feline herpesvirus type 1. Vet. Ophthalmol. 2016, 19 (Suppl. 1) (Suppl. 1), 119–130. [Google Scholar] [CrossRef] [Green Version]
- Posada, D. Using MODELTEST and PAUP* to select a model of nucleotide substitution. Curr. Protoc. Bioinform. 2003. [Google Scholar] [CrossRef]
- Kolb, A.W.; Larsen, I.V.; Cuellar, J.A.; Brandt, C.R. Genomic, phylogenetic, and recombinational characterization of herpes simplex virus 2 strains. J. Virol. 2015, 89, 6427–6434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, A.W.; Adams, M.; Cabot, E.L.; Craven, M.; Brandt, C.R. Multiplex sequencing of seven ocular herpes simplex virus type-1 genomes: Phylogeny, sequence variability, and SNP distribution. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9061–9073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szpara, M.L.; Parsons, L.; Enquist, L.W. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J. Virol. 2010, 84, 5303–5313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spertus, C.B.; Mohammed, H.O.; Ledbetter, E.C. Effects of topical ocular application of 1% trifluridine ophthalmic solution in dogs with experimentally induced recurrent ocular canine herpesvirus-1 infection. Am. J. Vet. Res. 2016, 77, 1140–1147. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.; Boivin, G. Antiviral resistance in herpes simplex virus and varicella-zoster virus infections: Diagnosis and management. Curr. Opin. Infect. Dis. 2016, 29, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kolb, A.W.; Sverchkov, Y.; Cuellar, J.A.; Craven, M.; Brandt, C.R. Recombination Analysis of Herpes Simplex Virus 1 Reveals a Bias toward GC Content and the Inverted Repeat Regions. J. Virol. 2015, 89, 7214–7223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohner, D.E.; Adams, S.G.; Gelb, L.D. Recombination in tissue culture between varicella-zoster virus strains. J. Med. Virol. 1988, 24, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Henderson, L.M.; Katz, J.B.; Erickson, G.A.; Mayfield, J.E. In vivo and in vitro genetic recombination between conventional and gene-deleted vaccine strains of pseudorabies virus. Am. J. Vet. Res. 1990, 51, 1656–1662. [Google Scholar]
- Miyoshi, M.; Ishii, Y.; Takiguchi, M.; Takada, A.; Yasuda, J.; Hashimoto, A.; Okazaki, K.; Kida, H. Detection of canine herpesvirus DNA in the ganglionic neurons and the lymph node lymphocytes of latently infected dogs. J. Vet. Med. Sci. 1999, 61, 375–379. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain ID | Genbank Accession | Host Age/Sex/Breed | Host Location | Date Collected | Source Sample |
|---|---|---|---|---|---|
| ELAL-1 | MW353125 | 7/FS/English Bulldog | San Antonio, TX, USA | 6 March 2019 | Eye |
| ELAL-2 | MW353126 | 8/MC/Labrador Retriever | Oneonta, NY, USA | 6 July 2003 | Eye |
| ELAL-3 | MW353127 | 0.33/F/Beagle | North Rose, NY, USA | 19 November 2008 | Eye |
| ELAL-4 | MW353128 | 0.25/F/Beagle | North Rose, NY, USA | 19 November 2008 | Eye |
| ELAL-5 | MW353129 | 7/FS/Min. Schnauzer | Endwell, NY, USA | 26 August 2004 | Eye |
| ELAL-6 | MW353130 | 10/FS/Golden Retriever | Williston, VT, USA | 17 September 2014 | Eye |
| ELAL-7 | MW353131 | 8/FS/Cross breed | Batavia, NY, USA | 18 October 2007 | Eye |
| ELAL-8 | MW353132 | 8/MC/ACS | Ovid, NY, USA | 25 February 2014 | Eye |
| ELAL-9 | MW353133 | 0.2/M/Beagle | North Rose, NY, USA | 19 November 2008 | Eye |
| ELAL-10 | MW353134 | 0.2/M/Beagle | North Rose, NY, USA | 19 November 2008 | Eye |
| ELAL-11 | MW353135 | 0.33/F/Beagle | North Rose, NY, USA | 19 November 2008 | Eye |
| ELAL-12 | MW353136 | 12/MC/Cross breed | Wilton, CT, USA | 16 October 2019 | Eye |
| ELAL-13 | MW353138 | 0.16/F/Beagle | Ithaca, NY, USA | 16 April 2009 | Eye |
| ELAL-14 | MW353137 | 0.33/M/Beagle | North Rose, NY, USA | 19 November 2008 | Eye |
| ELAL-15 | MW353139 | 9/FS/Golden Retriever | Binghamton, NY, USA | 26 August 2008 | Eye |
| V1154 | KT819631 | 0.04/NA/Dalmatian | UK | 2000 | Kidney |
| V777 | KT819632 | 0.04/NA/Min. Schnauzer | UK | 1995 | Lung |
| BTU-1 | KX828242 | NA/NA/NA | Brazil | 2012 | Kidney |
| 15-4016-NSW | KY057364 | 0.08/F/Labrador Retriever | NSW, Australia | 2016 | Liver |
| 0-194 | NC_030117 | NA/NA/NA | NA | 1985 | NA |
| C-27 | NC_013590 | NA/NA/NA | NA | Pre 1980 | NA |
| Strain ID | Number Reads | Mapped Reads | Mean Coverage | Mean Mapped Read Length (bp) |
|---|---|---|---|---|
| ELAL-1 | 2,865,972 | 155,016 | 290.2 | 226.9 |
| ELAL-2 | 2,963,230 | 390,228 | 706.8 | 228.6 |
| ELAL-3 | 1,292,262 | 231,149 | 413.2 | 225.3 |
| ELAL-4 | 1,848,000 | 376,806 | 663.9 | 223.1 |
| ELAL-5 | 1,430,708 | 279,714 | 476.8 | 214.7 |
| ELAL-6 | 3,053,006 | 847,371 | 1509.6 | 225.4 |
| ELAL-7 | 3,221,498 | 219,157 | 372.8 | 214.5 |
| ELAL-8 | 1,889,752 | 298,155 | 505.4 | 213.2 |
| ELAL-9 | 3,415,468 | 416,370 | 706 | 213.7 |
| ELAL-10 | 3,533,658 | 648,209 | 1409.8 | 201.2 |
| ELAL-11 | 2,356,364 | 456,166 | 765.2 | 211.7 |
| ELAL-12 | 3,126,278 | 639,677 | 1024.4 | 201.1 |
| ELAL-13 | 2,496,354 | 326,583 | 536.8 | 206.8 |
| ELAL-14 | 2,955,844 | 856,992 | 1404 | 207.5 |
| ELAL-15 | 2,985,658 | 527,658 | 870.8 | 205.7 |
| Gene | Gene Product | Number Unique Synonymous Variants | Number Unique non-Synonymous Variants | Length of Gene (bp) |
|---|---|---|---|---|
| Circ | Myristylated tegument protein CIRC | 4 | 2 | 651 |
| RL2 | Ubiquitin E3 ligase ICP0 | 1 | 4 | 1005 |
| RS1 | Transcriptional regulator ICP4 | 22 | 16 | 4155(F)/4155(R) |
| UL1 | Envelope glycoprotein L | 0 | 0 | 468 |
| UL10 | Envelope glycoprotein M | 1 | 2 | 1254 |
| UL11 | Myristylated tegument protein | 1 | 0 | 213 |
| UL12 | Deoxyribonuclease | 2 | 0 | 1638 |
| UL13 | Tegument serine/threonine protein kinase | 2 | 5 | 1770 |
| UL14 | Tegument protein UL14 | 1 | 4 | 912 |
| UL15 | DNA packaging terminase subunit 1 | 1 | 2 | 5442 |
| UL16 | Tegument protein UL16 | 4 | 2 | 1092 |
| UL17 | DNA packaging tegument protein UL17 | 0 | 4 | 2067 |
| UL18 | Capsid triplex subunit 2 | 3 | 0 | 948 |
| UL19 | Major capsid protein | 7 | 3 | 4122 |
| UL2 | Uracil-DNA glycosylase | 0 | 0 | 837 |
| UL20 | Envelope protein UL20 | 2 | 0 | 693 |
| UL21 | Tegument protein UL21 | 2 | 1 | 1572 |
| UL22 | Envelope glycoprotein H | 1 | 1 | 2394 |
| UL23 | Thymidine kinase | 0 | 2 | 987 |
| UL24 | Nuclear protein UL24 | 1 | 0 | 783 |
| UL25 | DNA packaging tegument protein UL25 | 2 | 1 | 1758 |
| UL26 | Capsid maturation protease | 4 | 2 | 1731 |
| UL26.5 | Capsid scaffold protein | 3 | 1 | 846 |
| UL27 | Envelope glycoprotein B | 1 | 1 | 2640 |
| UL28 | DNA packaging terminase subunit 2 | 3 | 2 | 2271 |
| UL29 | Single-stranded DNA-binding protein | 5 | 4 | 3594 |
| UL3 | Nuclear protein UL3 | 0 | 1 | 597 |
| UL30 | DNA polymerase catalytic subunit | 2 | 4 | 3540 |
| UL31 | Nuclear egress lamina protein | 1 | 1 | 1005 |
| UL32 | DNA packaging protein UL32 | 1 | 2 | 1680 |
| UL33 | DNA packaging protein UL33 | 0 | 1 | 381 |
| UL34 | Nuclear egress membrane protein | 1 | 1 | 795 |
| UL35 | Small capsid protein | 0 | 0 | 321 |
| UL36 | Large tegument protein | 15 | 42 | 9531 |
| UL37 | Tegument protein UL37 | 3 | 4 | 3039 |
| UL38 | Capsid triplex subunit 1 | 1 | 0 | 1374 |
| UL4 | Nuclear protein UL4 | 3 | 1 | 663 |
| UL41 | Tegument host shutoff protein | 0 | 4 | 1461 |
| UL42 | DNA polymerase processivity subunit | 1 | 2 | 1059 |
| UL43 | Envelope protein UL43 | 3 | 2 | 1203 |
| UL44 | Envelope glycoprotein C | 1 | 1 | 1380 |
| UL45 | Membrane protein UL45 | 1 | 1 | 528 |
| UL46 | Tegument protein VP11/12 | 0 | 1 | 1986 |
| UL47 | Tegument protein VP13/14 | 7 | 8 | 2439 |
| UL48 | Transactivating tegument protein VP16 | 3 | 1 | 1269 |
| UL49 | Tegument protein VP22 | 7 | 1 | 795 |
| UL49A | Envelope glycoprotein N | 3 | 1 | 261 |
| UL5 | Helicase-primase helicase subunit | 1 | 3 | 2661 |
| UL50 | Deoxyuridine triphosphate | 20 | 33 | 918 |
| UL51 | Tegument protein UL51 | 14 | 6 | 672 |
| UL52 | Helicase-primase primase subunit | 17 | 14 | 3063 |
| UL53 | Envelope glycoprotein K | 8 | 4 | 1014 |
| UL54 | Multifunctional expression regulator | 4 | 0 | 1272 |
| UL55 | Nuclear protein UL55 | 3 | 1 | 564 |
| UL56 | Membrane protein UL56 | 0 | 5 | 522 |
| UL6 | Capsid portal protein | 8 | 1 | 2073 |
| UL7 | Tegument protein UL7 | 2 | 2 | 879 |
| UL8 | Helicase-primase subunit | 6 | 3 | 2271 |
| UL9 | DNA replication origin-binding helicase | 3 | 3 | 2535 |
| US1 | Regulatory protein ICP22 | 5 | 1 | 942(F)/942(R) |
| US10 | Virion protein US10 | 1 | 5 | 588(F)/588(R) |
| US2 | Virion protein US2 | 3 | 2 | 1176 |
| US3 | Serine/threonine protein kinase US3 | 0 | 0 | 1203 |
| US4 | Envelope glycoprotein G | 1 | 1 | 1248 |
| US6 | Envelope glycoprotein D | 3 | 1 | 1038 |
| US7 | Envelope glycoprotein I | 3 | 1 | 1038 |
| US8 | Envelope glycoprotein E | 2 | 4 | 1569 |
| US8A | Membrane protein US8A | 0 | 0 | 237 |
| US9 | Membrane protein US9 | 0 | 2 | 360 |
| V32 | Protein V32 | 0 | 2 | 417 |
| V57 | Protein V57 | 1 | 1 | 729 |
| V67 | Virion protein V67 | 6 | 0 | 552(F)/552(R) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewin, A.C.; Coghill, L.M.; Mironovich, M.; Liu, C.-C.; Carter, R.T.; Ledbetter, E.C. Phylogenomic Analysis of Global Isolates of Canid Alphaherpesvirus 1. Viruses 2020, 12, 1421. https://doi.org/10.3390/v12121421
Lewin AC, Coghill LM, Mironovich M, Liu C-C, Carter RT, Ledbetter EC. Phylogenomic Analysis of Global Isolates of Canid Alphaherpesvirus 1. Viruses. 2020; 12(12):1421. https://doi.org/10.3390/v12121421
Chicago/Turabian StyleLewin, Andrew C., Lyndon M. Coghill, Melanie Mironovich, Chin-Chi Liu, Renee T. Carter, and Eric C. Ledbetter. 2020. "Phylogenomic Analysis of Global Isolates of Canid Alphaherpesvirus 1" Viruses 12, no. 12: 1421. https://doi.org/10.3390/v12121421
APA StyleLewin, A. C., Coghill, L. M., Mironovich, M., Liu, C.-C., Carter, R. T., & Ledbetter, E. C. (2020). Phylogenomic Analysis of Global Isolates of Canid Alphaherpesvirus 1. Viruses, 12(12), 1421. https://doi.org/10.3390/v12121421