Recombination and Positive Selection Differentially Shaped the Diversity of Betacoronavirus Subgenera




Abstract
1. Introduction
2. Materials and Methods
2.1. Sequences and Alignments
2.2. Recombination Analysis
2.3. Detection of Positive Selection in Betacoronavirus Subgenera
3. Results
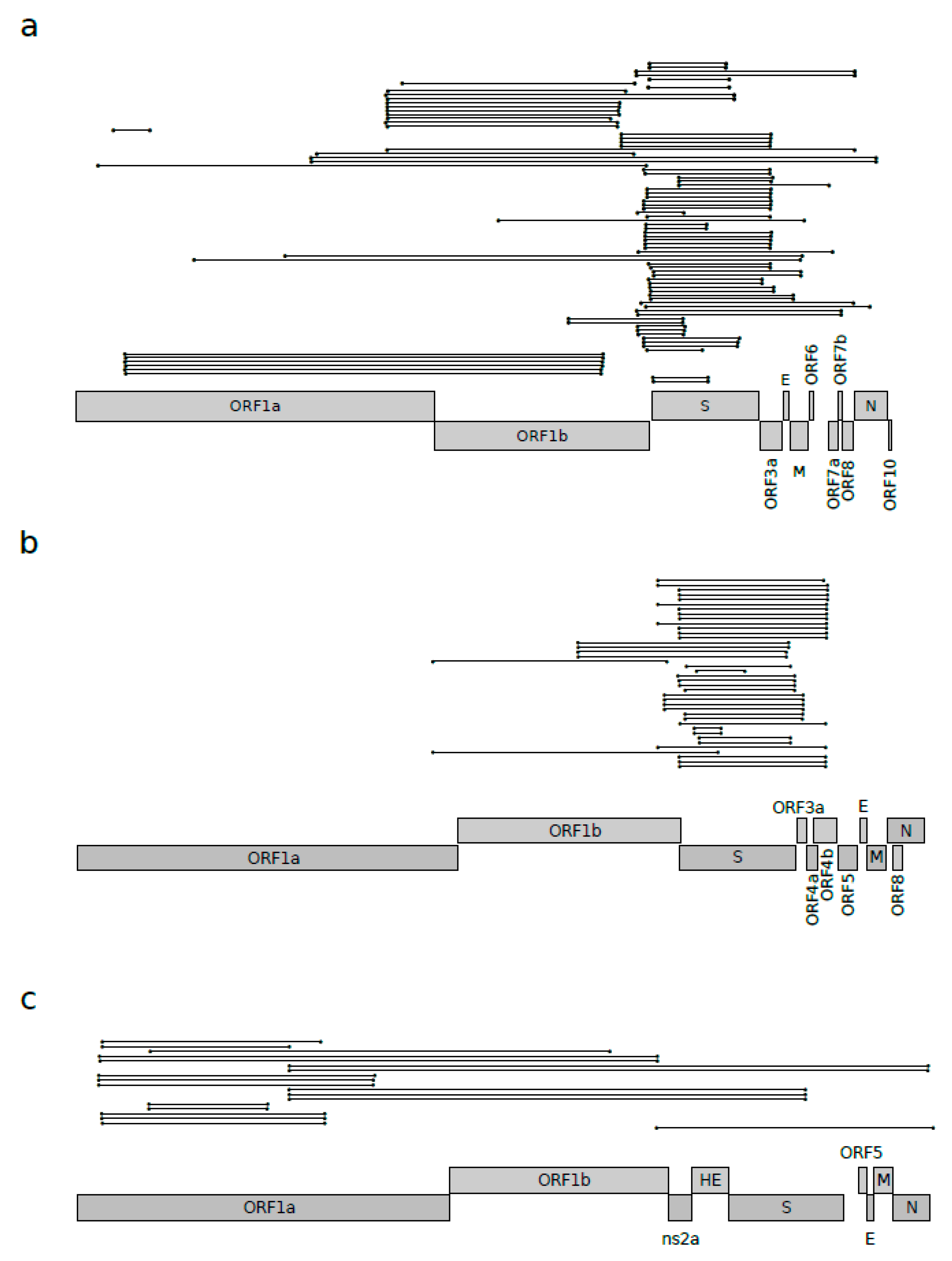
3.1. Recombination Plays a Major Role in Sarbecovirus Evolution
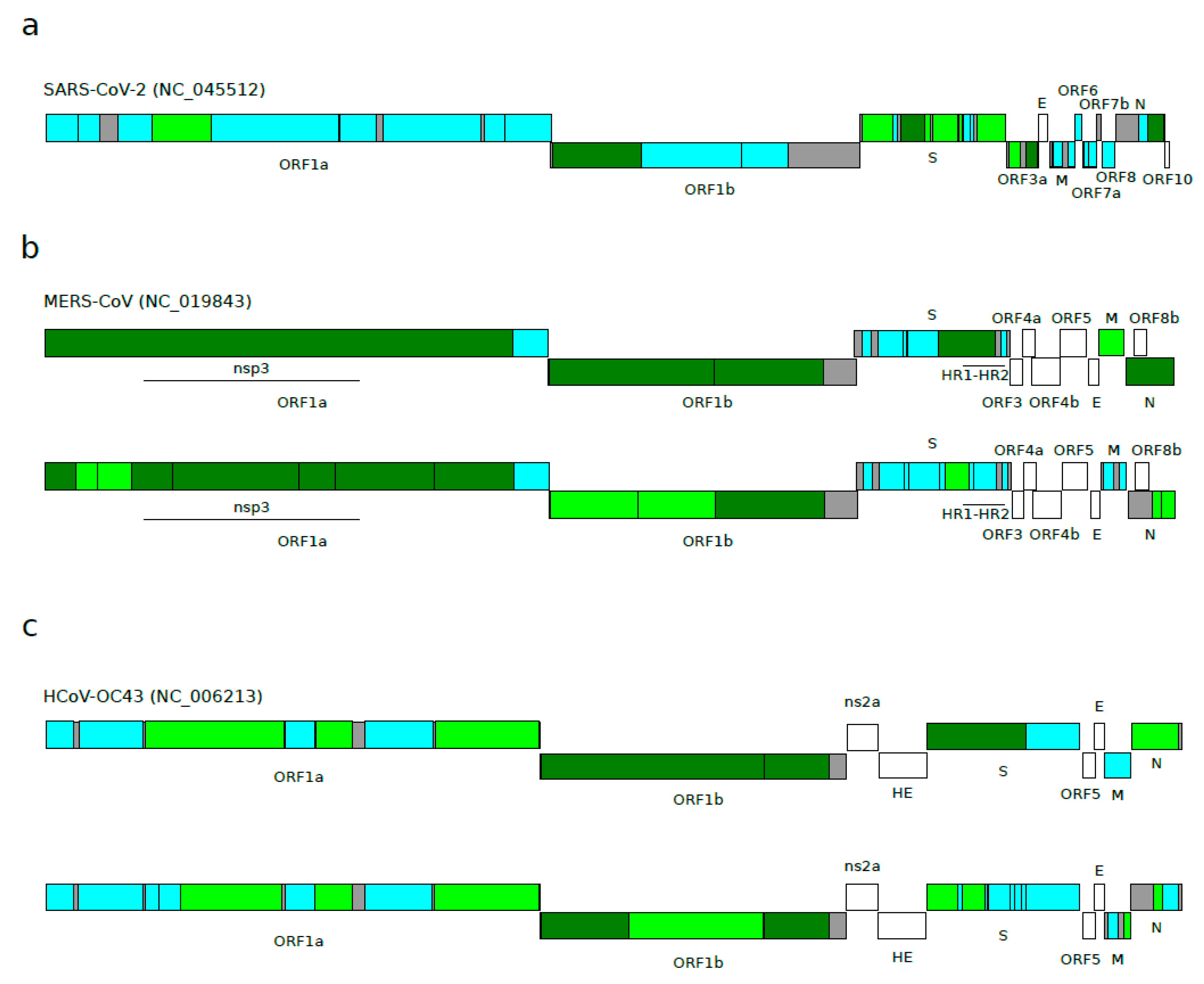
3.2. Positive Selection Acting on Betacoronaviruses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Luk, H.K.H.; Li, X.; Fung, J.; Lau, S.K.P.; Woo, P.C.Y. Molecular Epidemiology, Evolution and Phylogeny of SARS Coronavirus. Infect. Genet. Evol. 2019, 71, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.L. Origin and Evolution of Pathogenic Coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef]
- de Groot, R.J.; Baker, S.C.; Baric, R.S.; Brown, C.S.; Drosten, C.; Enjuanes, L.; Fouchier, R.A.; Galiano, M.; Gorbalenya, A.E.; Memish, Z.A.; et al. Middle East Respiratory Syndrome Coronavirus (MERS-CoV): Announcement of the Coronavirus Study Group. J. Virol. 2013, 87, 7790–7792. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Snijder, E.J.; Spaan, W.J. Severe Acute Respiratory Syndrome Coronavirus Phylogeny: Toward Consensus. J. Virol. 2004, 78, 7863–7866. [Google Scholar] [CrossRef] [PubMed]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-nCoV and Naming it SARS-CoV-2. Nat. Microbiol 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Killerby, M.E.; Biggs, H.M.; Midgley, C.M.; Gerber, S.I.; Watson, J.T. Middle East Respiratory Syndrome Coronavirus Transmission. Emerg. Infect. Dis. 2020, 26, 191–198. [Google Scholar] [CrossRef]
- Lam, T.T.; Shum, M.H.; Zhu, H.; Tong, Y.; Ni, X.; Liao, Y.; Wei, W.; Cheung, W.Y.; Li, W.; Li, L.; et al. Identification of 2019-nCoV Related Coronaviruses in Malayan Pangolins in Southern China. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef]
- Xiao, K.; Zhai, J.; Feng, Y.; Zhou, N.; Zhang, X.; Zou, J.; Li, N.; Guo, Y.; Li, X.; Shen, X.; et al. Isolation and Characterization of 2019-nCoV-Like Coronavirus from Malayan Pangolins. BioRxiv 2020. [Google Scholar] [CrossRef]
- Wong, M.C.; Javornik Cregeen, S.J.; Ajami, N.J.; Petrosino, J.F. Evidence of Recombination in Coronaviruses Implicating Pangolin Origins of nCoV-2019. BioRxiv 2020. [Google Scholar] [CrossRef]
- Liu, P.; Jiang, J.; Wan, X.; Hua, Y.; Wang, X.; Hou, F.; Chen, J.; Zou, J.; Chen, J. Are Pangolins the Intermediate Host of the 2019 Novel Coronavirus (2019-nCoV)? BioRxiv 2020. [Google Scholar] [CrossRef]
- Ye, Z.W.; Yuan, S.; Yuen, K.S.; Fung, S.Y.; Chan, C.P.; Jin, D.Y. Zoonotic Origins of Human Coronaviruses. Int. J. Biol. Sci. 2020, 16, 1686–1697. [Google Scholar] [CrossRef]
- Graham, R.L.; Baric, R.S. Recombination, Reservoirs, and the Modular Spike: Mechanisms of Coronavirus Cross-Species Transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef]
- Dudas, G.; Rambaut, A. MERS-CoV recombination: Implications about the reservoir and potential for adaptation. Virus Evol. 2016, 2, vev023. [Google Scholar] [CrossRef]
- Hon, C.C.; Lam, T.Y.; Shi, Z.L.; Drummond, A.J.; Yip, C.W.; Zeng, F.; Lam, P.Y.; Leung, F.C. Evidence of the Recombinant Origin of a Bat Severe Acute Respiratory Syndrome (SARS)-Like Coronavirus and its Implications on the Direct Ancestor of SARS Coronavirus. J. Virol. 2008, 82, 1819–1826. [Google Scholar] [CrossRef]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary Origins of the SARS-CoV-2 Sarbecovirus Lineage Responsible for the COVID-19 Pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef]
- Simmonds, P.; Aiewsakun, P.; Katzourakis, A. Prisoners of War—Host Adaptation and its Constraints on Virus Evolution. Nat. Rev. Microbiol. 2019, 17, 321–328. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Lam, H.M.; Ratmann, O.; Boni, M.F. Improved Algorithmic Complexity for the 3SEQ Recombination Detection Algorithm. Mol. Biol. Evol. 2018, 35, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Cagliani, R.; Forni, D.; Clerici, M.; Sironi, M. Coding Potential and Sequence Conservation of SARS-CoV-2 and Related Animal Viruses. Infect. Genet. Evol. 2020, 83, 104353. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Delsuc, F.; Dufayard, J.F.; Gascuel, O. Estimating Maximum Likelihood Phylogenies with PhyML. Methods Mol. Biol. 2009, 537, 113–137. [Google Scholar] [PubMed]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Kosakovsky Pond, S.L. Less is More: An Adaptive Branch-Site Random Effects Model for Efficient Detection of Episodic Diversifying Selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.; Frost, S.D.; Muse, S.V. HyPhy: Hypothesis Testing using Phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Lemey, P.; Posada, D. Analysing Recombination in Nucleotide Sequences. Mol. Ecol. Resour. 2011, 11, 943–955. [Google Scholar] [CrossRef]
- Lau, S.K.; Feng, Y.; Chen, H.; Luk, H.K.; Yang, W.H.; Li, K.S.; Zhang, Y.Z.; Huang, Y.; Song, Z.Z.; Chow, W.N.; et al. Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination. J. Virol. 2015, 89, 10532–10547. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, L.; Ren, X.; Zhang, J.; Yang, F.; Zhang, S.; Jin, Q. ORF8-Related Genetic Evidence for Chinese Horseshoe Bats as the Source of Human Severe Acute Respiratory Syndrome Coronavirus. J. Infect. Dis. 2016, 213, 579–583. [Google Scholar] [CrossRef]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a Rich Gene Pool of Bat SARS-Related Coronaviruses Provides New Insights into the Origin of SARS Coronavirus. PLoS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef]
- Cotten, M.; Watson, S.J.; Zumla, A.I.; Makhdoom, H.Q.; Palser, A.L.; Ong, S.H.; Al Rabeeah, A.A.; Alhakeem, R.F.; Assiri, A.; Al-Tawfiq, J.A.; et al. Spread, Circulation, and Evolution of the Middle East Respiratory Syndrome Coronavirus. MBio 2014, 5. [Google Scholar] [CrossRef]
- Li, X.; Giorgi, E.E.; Marichannegowda, M.H.; Foley, B.; Xiao, C.; Kong, X.-P.; Chen, Y.; Gnanakaran, S.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv. 2020, 6, eabb9153. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Shafique, L.; Ihsan, A.; Liu, Q. Evolutionary Trajectory for the Emergence of Novel Coronavirus SARS-CoV-2. Pathogens 2020, 9, 240. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; van der Walt, E.; Posada, D.; Rybicki, E.P. The Evolutionary Value of Recombination is Constrained by Genome Modularity. PLoS Genet. 2005, 1, e51. [Google Scholar] [CrossRef] [PubMed]
- Galli, A.; Kearney, M.; Nikolaitchik, O.A.; Yu, S.; Chin, M.P.; Maldarelli, F.; Coffin, J.M.; Pathak, V.K.; Hu, W.S. Patterns of Human Immunodeficiency Virus Type 1 Recombination Ex Vivo Provide Evidence for Coadaptation of Distant Sites, Resulting in Purifying Selection for Intersubtype Recombinants during Replication. J. Virol. 2010, 84, 7651–7661. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Rybicki, E.P. Investigation of Maize Streak Virus Pathogenicity Determinants using Chimaeric Genomes. Virology 2002, 300, 180–188. [Google Scholar] [CrossRef]
- Simon-Loriere, E.; Galetto, R.; Hamoudi, M.; Archer, J.; Lefeuvre, P.; Martin, D.P.; Robertson, D.L.; Negroni, M. Molecular Mechanisms of Recombination Restriction in the Envelope Gene of the Human Immunodeficiency Virus. PLoS Pathog. 2009, 5, e1000418. [Google Scholar] [CrossRef]
- Monjane, A.L.; Martin, D.P.; Lakay, F.; Muhire, B.M.; Pande, D.; Varsani, A.; Harkins, G.; Shepherd, D.N.; Rybicki, E.P. Extensive Recombination-Induced Disruption of Genetic Interactions is Highly Deleterious but can be Partially Reversed by Small Numbers of Secondary Recombination Events. J. Virol. 2014, 88, 7843–7851. [Google Scholar] [CrossRef]
- Archer, J.; Pinney, J.W.; Fan, J.; Simon-Loriere, E.; Arts, E.J.; Negroni, M.; Robertson, D.L. Identifying the Important HIV-1 Recombination Breakpoints. PLoS Comput. Biol. 2008, 4, e1000178. [Google Scholar] [CrossRef]
- Lefeuvre, P.; Lett, J.M.; Varsani, A.; Martin, D.P. Widely Conserved Recombination Patterns among Single-Stranded DNA Viruses. J. Virol. 2009, 83, 2697–2707. [Google Scholar] [CrossRef]
- Bentley, K.; Evans, D.J. Mechanisms and Consequences of Positive-Strand RNA Virus Recombination. J. Gen. Virol. 2018, 99, 1345–1356. [Google Scholar] [CrossRef]
- Hoxie, I.; Dennehy, J.J. Intragenic Recombination Influences Rotavirus Diversity and Evolution. Virus Evol. 2020, 6, vez059. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Kosakovsky Pond, S.L. Purifying Selection can Obscure the Ancient Age of Viral Lineages. Mol. Biol. Evol. 2011, 28, 3355–3365. [Google Scholar] [CrossRef] [PubMed]
- Sironi, M.; Cagliani, R.; Forni, D.; Clerici, M. Evolutionary Insights into Host-Pathogen Interactions from Mammalian Sequence Data. Nat. Rev. Genet. 2015, 16, 224–236. [Google Scholar] [CrossRef] [PubMed]
- MacLean, O.A.; Lytras, S.; Weaver, S.; Singer, J.B.; Boni, M.F.; Lemey, P.; Kosakovsky Pond, S.L.; Robertson, D.L. Natural Selection in the Evolution of SARS-CoV-2 in Bats, Not Humans, Created a Highly Capable Human Pathogen. BioRxiv 2020. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Mozzi, A.; Pozzoli, U.; Al-Daghri, N.; Clerici, M.; Sironi, M. Extensive Positive Selection Drives the Evolution of Nonstructural Proteins in Lineage C Betacoronaviruses. J. Virol. 2016, 90, 3627–3639. [Google Scholar] [CrossRef]
- Gabriel, G.; Dauber, B.; Wolff, T.; Planz, O.; Klenk, H.D.; Stech, J. The Viral Polymerase Mediates Adaptation of an Avian Influenza Virus to a Mammalian Host. Proc. Natl. Acad. Sci. USA 2005, 102, 18590–18595. [Google Scholar] [CrossRef]
- Li, Z.; Chen, H.; Jiao, P.; Deng, G.; Tian, G.; Li, Y.; Hoffmann, E.; Webster, R.G.; Matsuoka, Y.; Yu, K. Molecular Basis of Replication of Duck H5N1 Influenza Viruses in a Mammalian Mouse Model. J. Virol. 2005, 79, 12058–12064. [Google Scholar] [CrossRef]
- Bhatt, S.; Lam, T.T.; Lycett, S.J.; Leigh Brown, A.J.; Bowden, T.A.; Holmes, E.C.; Guan, Y.; Wood, J.L.; Brown, I.H.; Kellam, P.; et al. The Evolutionary Dynamics of Influenza A Virus Adaptation to Mammalian Hosts. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2013, 368, 20120382. [Google Scholar] [CrossRef]
- Forni, D.; Filippi, G.; Cagliani, R.; De Gioia, L.; Pozzoli, U.; Al-Daghri, N.; Clerici, M.; Sironi, M. The Heptad Repeat Region is a Major Selection Target in MERS-CoV and Related Coronaviruses. Sci. Rep. 2015, 5, 14480. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Sarbecovirus | |||
|---|---|---|---|
| ORF | Region | Alignment Length | aBSREL Result |
| ORF1a | |||
| reg1 | 834 | 0 branches under selection among 43 tested | |
| reg2 | 570 | 0 branches under selection among 43 tested | |
| reg3 | 897 | 0 branches under selection among 43 tested | |
| reg4 | 1653 | 1 branch under selection among 43 tested | |
| reg5 | 3393 | 0 branches under selection among 43 tested | |
| reg6 | 963 | 0 branches under selection among 43 tested | |
| reg7 | 2568 | 0 branches under selection among 43 tested | |
| reg8 | 537 | 0 branches under selection among 43 tested | |
| reg9 | 1221 | 0 branches under selection among 43 tested | |
| ORF1b | |||
| reg1 | 2316 | 2 branches under selection among 43 tested. | |
| reg2 | 2613 | 0 branches under selection among 43 tested | |
| reg3 | 1212 | 0 branches under selection among 43 tested | |
| S | |||
| reg1 | 843 | 1 branch under selection among 43 tested | |
| reg2 | 141 | 0 branches under selection among 43 tested | |
| reg3 | 624 | 2 branches under selection among 43 tested | |
| reg4 | 141 | 1 branch under selection among 42 tested | |
| reg5 | 648 | 1 branch under selection among 43 tested | |
| reg6 | 114 | 1 branch under selection among 43 tested | |
| reg7 | 183 | 0 branches under selection among 43 tested | |
| reg8 | 114 | 0 branches under selection among 43 tested | |
| reg9 | 750 | 1 branch under selection among 43 tested | |
| ORF3a | |||
| reg1 | 291 | 1 branch under selection among 43 tested | |
| reg2 | 315 | 3 branches under selection among 43 tested | |
| M | |||
| reg1 | 261 | 0 branches under selection among 43 tested | |
| reg2 | 171 | 0 branches under selection among 42 tested | |
| ORF6 | 183 | 0 branches under selection among 42 tested | |
| ORF7a | |||
| reg1 | 126 | 0 branches under selection among 42 tested | |
| reg2 | 216 | 0 branches under selection among 43 tested | |
| ORF8 | |||
| reg1 | 378 | 0 branches under selection among 40 tested | |
| N | |||
| reg1 | 234 | 0 branches under selection among 41 tested | |
| reg2 | 444 | 2 branches under selection among 43 tested | |
| ORF | Region | Alignment Length | aBSREL Result |
|---|---|---|---|
| ORF1a | |||
| reg1 | 12,960 | 7 branches under selection among 12 tested | |
| reg2 | 927 | 0 branches under selection among 12 tested | |
| ORF1b | |||
| reg1 | 4314 | 4 branches under selection among 12 tested | |
| reg2 | 2889 | 3 branches under selection among 12 tested | |
| S | |||
| reg1 | 279 | 0 branches under selection among 12 tested | |
| reg2 | 654 | 0 branches under selection among 12 tested | |
| reg3 | 108 | 0 branches under selection among 12 tested | |
| reg4 | 849 | 0 branches under selection among 12 tested | |
| reg5 | 1512 | 3 branches under selection among 12 tested | |
| reg6 | 150 | 0 branches under selection among 12 tested | |
| M | 663 | 1 branch under selection among 12 tested | |
| N | 1341 | 6 branches under selection among 12 tested |
| ORF | Region | Alignment Length | aBSREL Result |
|---|---|---|---|
| ORF1a | |||
| reg1 | 723 | 0 branches under selection among 12 tested | |
| reg2 | 1731 | 0 branches under selection among 12 tested | |
| reg3 | 4419 | 1 branch under selection among 12 tested | |
| reg4 | 783 | 0 branches under selection among 12 tested | |
| reg5 | 987 | 1 branch under selection among 12 tested | |
| reg6 | 1800 | 0 branches under selection among 12 tested | |
| reg7 | 2775 | 1 branch under selection among 12 tested | |
| ORF1b | |||
| reg1 | 5919 | 3 branches under selection among 12 tested | |
| reg2 | 1740 | 3 branches under selection among 12 tested | |
| S | |||
| reg1 | 2823 | 2 branches under selection among 12 tested | |
| reg2 | 1449 | 0 branches under selection among 12 tested | |
| M | 693 | 0 branches under selection among 12 tested | |
| N | reg1 | 1299 | 1 branch under selection among 12 tested |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forni, D.; Cagliani, R.; Sironi, M. Recombination and Positive Selection Differentially Shaped the Diversity of Betacoronavirus Subgenera. Viruses 2020, 12, 1313. https://doi.org/10.3390/v12111313
Forni D, Cagliani R, Sironi M. Recombination and Positive Selection Differentially Shaped the Diversity of Betacoronavirus Subgenera. Viruses. 2020; 12(11):1313. https://doi.org/10.3390/v12111313
Chicago/Turabian StyleForni, Diego, Rachele Cagliani, and Manuela Sironi. 2020. "Recombination and Positive Selection Differentially Shaped the Diversity of Betacoronavirus Subgenera" Viruses 12, no. 11: 1313. https://doi.org/10.3390/v12111313
APA StyleForni, D., Cagliani, R., & Sironi, M. (2020). Recombination and Positive Selection Differentially Shaped the Diversity of Betacoronavirus Subgenera. Viruses, 12(11), 1313. https://doi.org/10.3390/v12111313