Simplified Bioprinting-Based 3D Cell Culture Infection Models for Virus Detection

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. 3D Bioprinting of Cultivation Matrices
2.3. Generation of 3D Cell Cultures on the Wellbricks Matrices
2.4. Virus Propagation, Purification, and Titration
2.5. Infection of 3D Cell Cultures
2.6. Cryo-Scanning Electron Microscopy (Cryo-SEM) of the Wellbrick Surface
2.7. Immunofluorescence Assay (IFA)
2.8. Live Cell Imaging of Infected 3D Cultures
2.9. DNA/RNA Preparation and Real-Time PCR Assays
2.10. Cell Viability Assay
3. Results
3.1. 3D Bioprinted Matrices Are Structurally Suitable for an Easy-to-Handle 3D Cell Cultivation
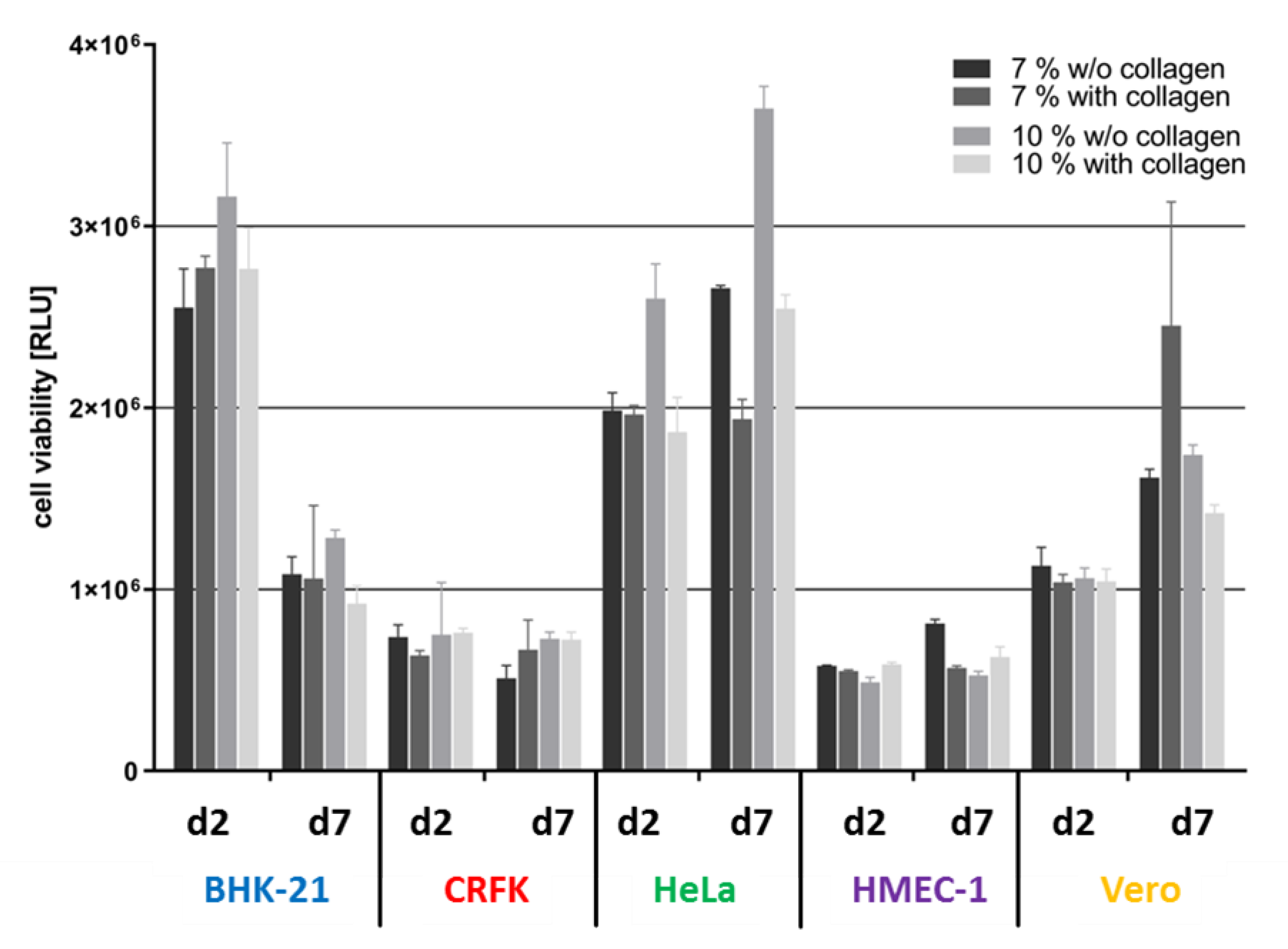
3.2. Matrix Composition Influences Viability of the Wellbricks Cell Cultures
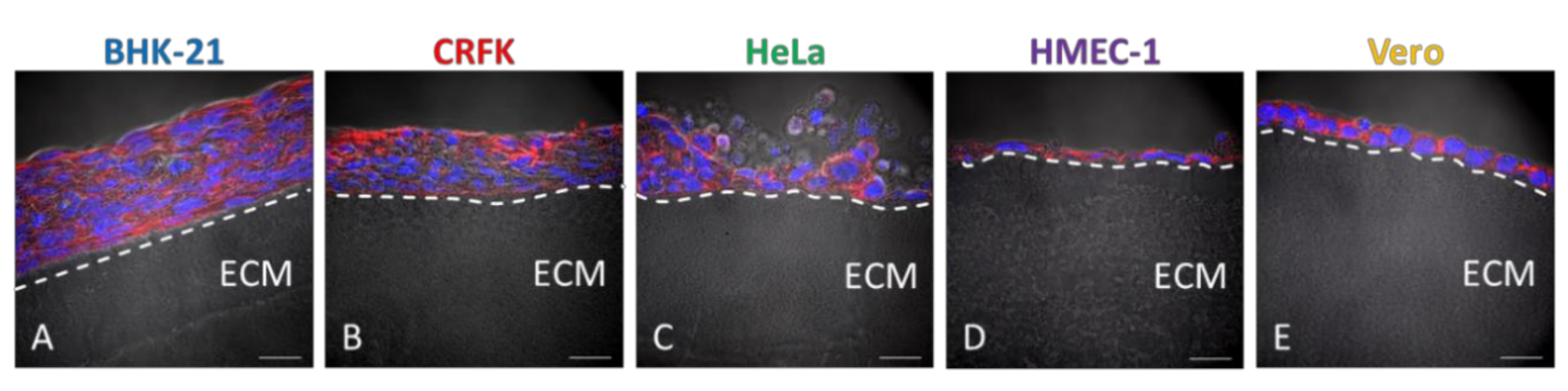
3.3. Cell lines Cultured on Wellbricks Show Distinct 3D-Typical Growth Characteristics
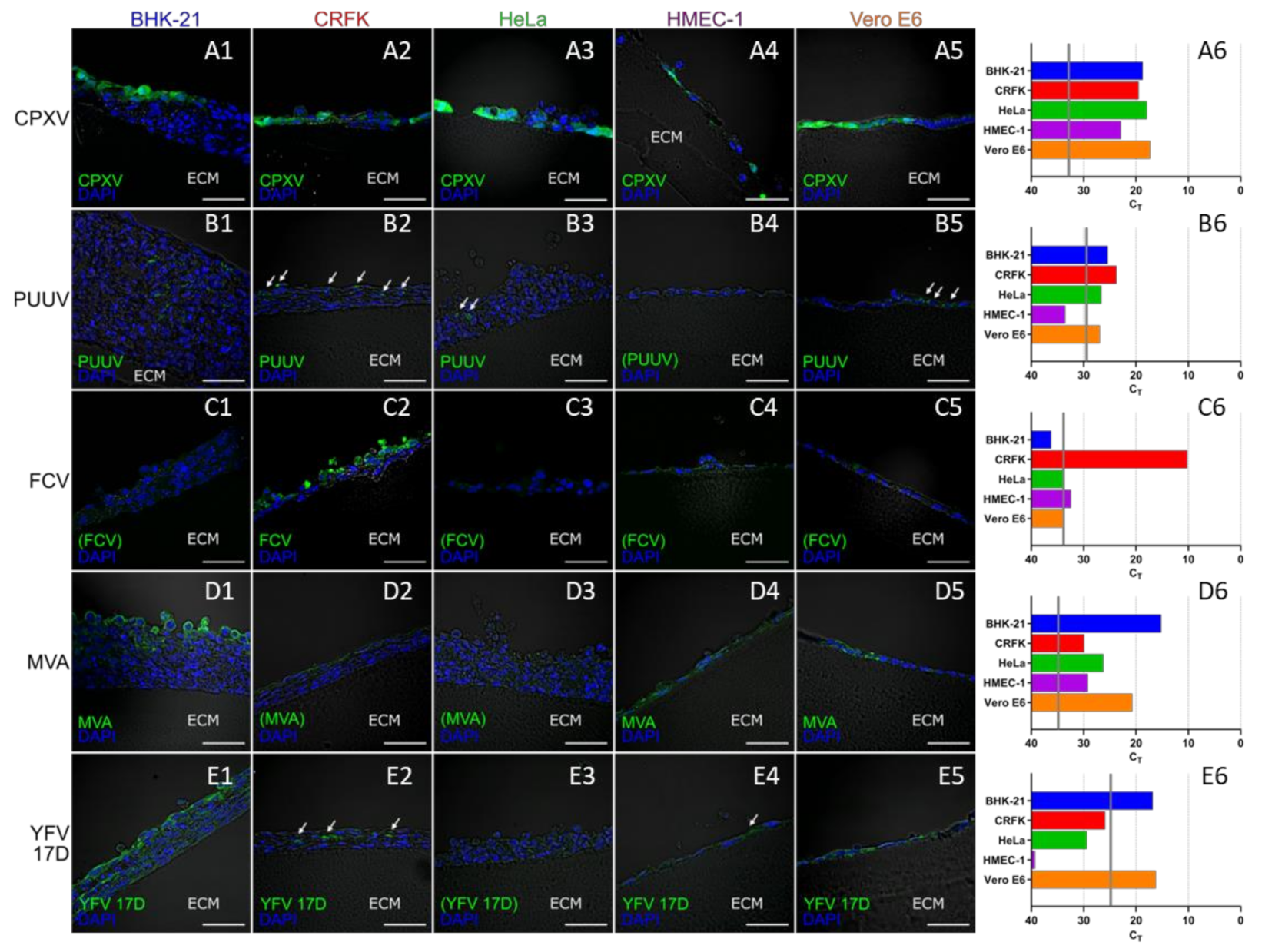
3.4. 3D-Cultured Cell Lines on Wellbricks Show Differential Permissiveness for the Infection with Different Viruses
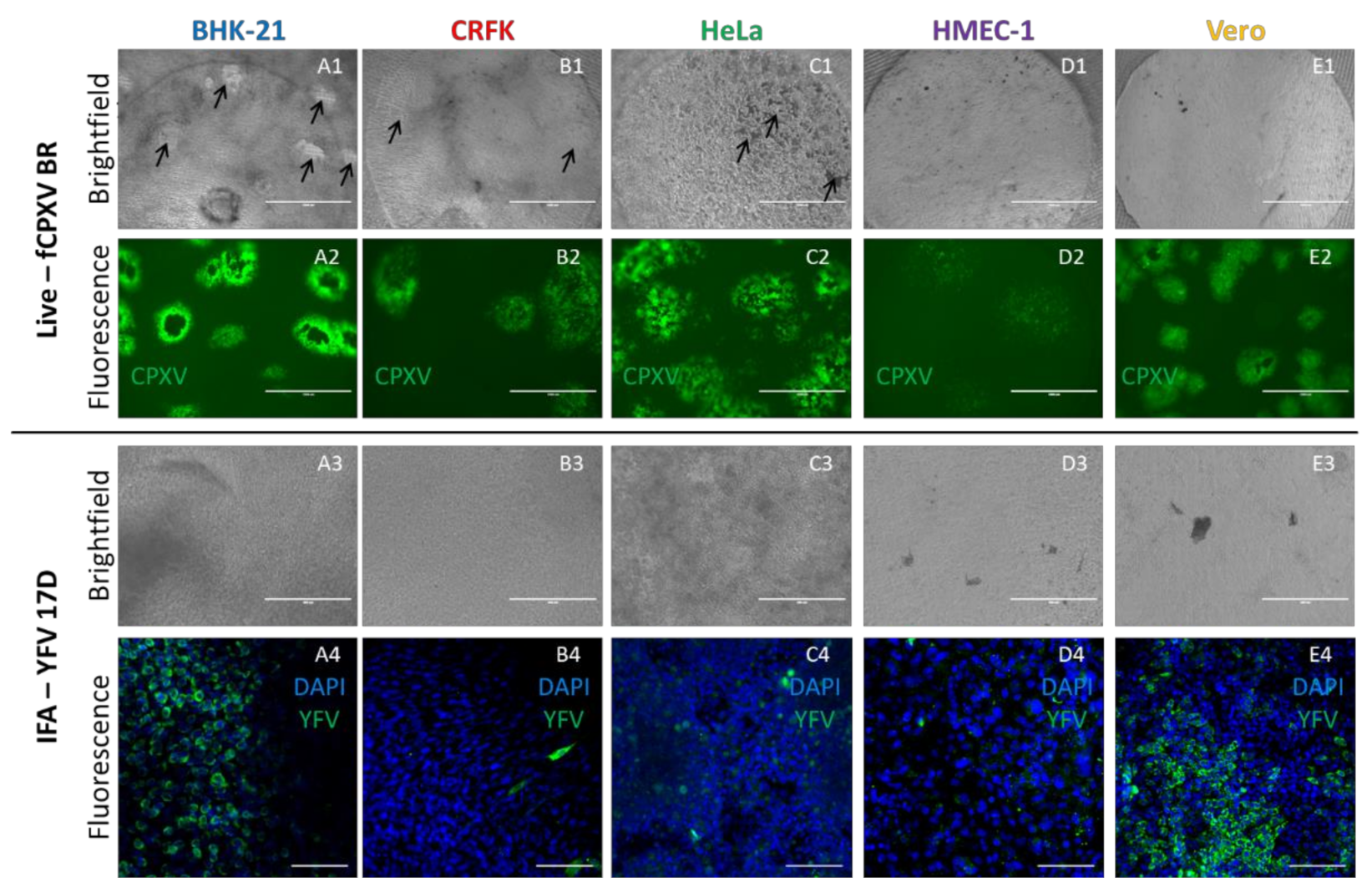
3.5. Screening of Infection in Wellbricks Cultures Is Simplified Compared to Other 3D Cell Culture Infection Models
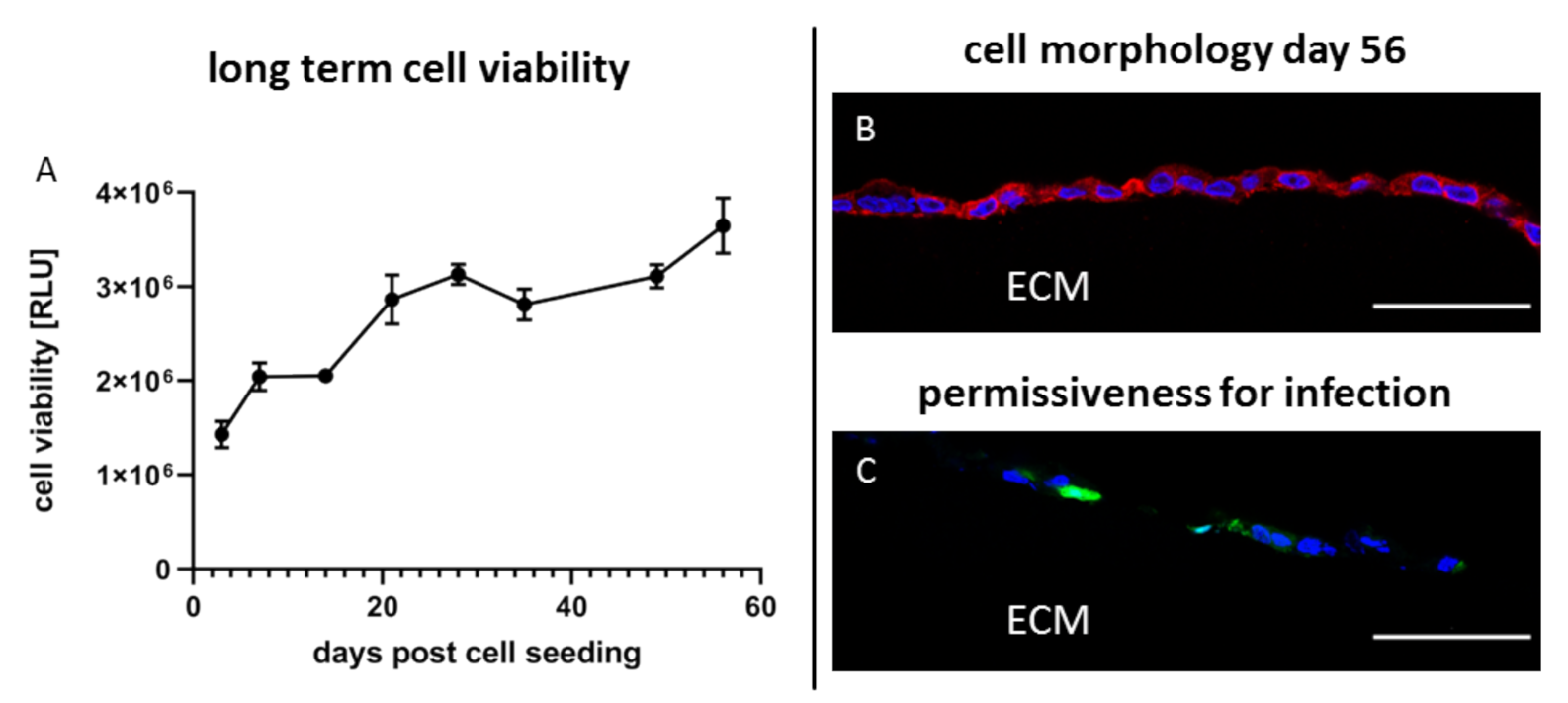
3.6. 3D Wellbricks Cultures Are Long-Term viable with Unaltered Permissiveness for Virus Infection
4. Discussion
4.1. 3D Bioprinted Matrices Promote In Vivo-Like Growth of Different Cell Types
4.2. Wellbricks 3D Cell Cultures Are Suitable for the In Vivo-Relevant Investigation of Viral Infections
4.3. Wellbricks 3D Cell Cultures Are Superior to Other 3D Cell Culture Models in Terms of Handling and Infection Screening
4.4. Wellbricks 3D Cell Cultures Represent a Suitable Tool for Virus Detection
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Species | Tissue | Cell Type | Origin * |
|---|---|---|---|---|
| BHK-21 | Mesocricetus auratus | Kidney | Fibroblast | ATCC; #CCL-10 |
| CRFK | Felis catus | Kidney | Epithelial | Friedrich Loeffler Institute; CCLV-RIE 769 |
| HeLa | Homo sapiens | Cervix | Epithelial | ATCC; #CCL-2 |
| HMEC-1 | Homo sapiens | Dermal | Endothelial | ATCC; #CRL-3243 |
| Vero E6 | Chlorocebus sabaeus | Kidney | Epithelial | ECACC; #85020206 |
| Virus * | Purification Buffer | Sucrose Cushion | Speed (× g) | Time (h) |
|---|---|---|---|---|
| FCV | 10 mM TrisHCl | 30% sucrose | 71,100 | 5 |
| MVA BN | 10 mM TrisHCl | 36% sucrose | 32,900 | 1.3 |
| PUUV Kazan | TNE buffer | 30% sucrose | 153,700 | 2 |
| YFV 17D | 10 mM TrisHCl | 36% sucrose | 153,700 | 5 |
| Feature/Virus | CPXV BR | MVA | FCV | PUUV | YFV 17D |
|---|---|---|---|---|---|
| Nucleic acid | dsDNA | dsDNA | ss(+)RNA | ss(−)RNA | ss(+)RNA |
| Replication rate (in vitro) | High | Intermediate | High | Low | Low |
| Virus genomes per cell (in vitro) | Intermediate | Intermediate | High | Low | High |
| Known host range (in vitro) | Broad | Intermediate | Narrow | Narrow | Intermediate |
| Cytopathogenic (in vitro) | Yes | Yes | Yes | No | No |
| Human pathogenic | Yes [60] | No [61] | No [62] | Yes [63] | No [64] |
| Surrogate for * | MPXV, VARV [65] | (MPXV, VARV) | NoV [66] | DOBV/HTNV [67] | YFV [64] |
References
- Enders, J.F.; Weller, T.H.; Robbins, F.C. Cultivation of the Lansing Strain of Poliomyelitis Virus in Cultures of Various Human Embryonic Tissues. Science 1949, 109, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Postlethwaite, R. Molluscum contagiosum. Arch. Environ. Health 1970, 21, 432–452. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. Model systems of human papillomavirus-associated disease. J. Pathol. 2016, 238, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Pfeiffer, J.K. Virology. Leaping the norovirus hurdle. Science 2014, 346, 700–701. [Google Scholar] [CrossRef]
- He, B.; Chen, G.; Zeng, Y. Three-dimensional cell culture models for investigating human viruses. Virol. Sin. 2016, 31, 363–379. [Google Scholar] [CrossRef]
- Koban, R.; Neumann, M.; Daugs, A.; Bloch, O.; Nitsche, A.; Langhammer, S.; Ellerbrok, H. A novel three-dimensional cell culture method enhances antiviral drug screening in primary human cells. Antivir. Res. 2018, 150, 20–29. [Google Scholar] [CrossRef]
- Andrei, G.; Duraffour, S.; Van den Oord, J.; Snoeck, R. Epithelial raft cultures for investigations of virus growth, pathogenesis and efficacy of antiviral agents. Antivir. Res. 2010, 85, 431–449. [Google Scholar] [CrossRef]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef]
- Osterman, A.; Stellberger, T.; Gebhardt, A.; Kurz, M.; Friedel, C.C.; Uetz, P.; Nitschko, H.; Baiker, A.; Vizoso-Pinto, M.G. The Hepatitis E virus intraviral interactome. Sci. Rep. 2015, 5, 13872. [Google Scholar] [CrossRef]
- Paneth Iheozor-Ejiofor, R.; Levanov, L.; Hepojoki, J.; Strandin, T.; Lundkvist, A.; Plyusnin, A.; Vapalahti, O. Vaccinia virus-free rescue of fluorescent replication-defective vesicular stomatitis virus and pseudotyping with Puumala virus glycoproteins for use in neutralization tests. J. Gen. Virol. 2016, 97, 1052–1059. [Google Scholar] [CrossRef]
- Yi, M.; Lemon, S.M. Replication of subgenomic hepatitis A virus RNAs expressing firefly luciferase is enhanced by mutations associated with adaptation of virus to growth in cultured cells. J. Virol. 2002, 76, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension - how 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Dollard, S.C.; Wilson, J.L.; Demeter, L.M.; Bonnez, W.; Reichman, R.C.; Broker, T.R.; Chow, L.T. Production of human papillomavirus and modulation of the infectious program in epithelial raft cultures. OFF. Genes Dev. 1992, 6, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.; Mane, M.; Kokorina, N.; Alam, S.; Hermonat, P.L. Ubiquitous human adeno-associated virus type 2 autonomously replicates in differentiating keratinocytes of a normal skin model. Virology 2000, 272, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Israr, M.; Mitchell, D.; Alam, S.; Dinello, D.; Kishel, J.J.; Meyers, C. Effect of the HIV protease inhibitor amprenavir on the growth and differentiation of primary gingival epithelium. Antivir. Ther. 2010, 15, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Abouleila, Y.; Si, L.; Ortega-Prieto, A.M.; Mummery, C.L.; Ingber, D.E.; Mashaghi, A. Human Organs-on-Chips for Virology. Trends Microbiol. 2020, 28, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Eglen, R.M. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov. 2017, 22, 456–472. [Google Scholar]
- Kacarevic, Z.P.; Rider, P.M.; Alkildani, S.; Retnasingh, S.; Smeets, R.; Jung, O.; Ivanisevic, Z.; Barbeck, M. An Introduction to 3D Bioprinting: Possibilities, Challenges and Future Aspects. Materials 2018, 11, 2199. [Google Scholar] [CrossRef]
- Jian, H.; Wang, M.; Wang, S.; Wang, A.; Bai, S. 3D bioprinting for cell culture and tissue fabrication. Bio-Design Manuf. 2018, 1, 45–61. [Google Scholar] [CrossRef]
- Grix, T.; Ruppelt, A.; Thomas, A.; Amler, A.K.; Noichl, B.P.; Lauster, R.; Kloke, L. Bioprinting Perfusion-Enabled Liver Equivalents for Advanced Organ-on-a-Chip Applications. Genes 2018, 9, 176. [Google Scholar] [CrossRef] [PubMed]
- Mazzocchi, A.; Soker, S.; Skardal, A. 3D bioprinting for high-throughput screening: Drug screening, disease modeling, and precision medicine applications. Appl. Phys. Rev. 2019, 6, 011302. [Google Scholar] [CrossRef]
- Kelsey, R.; Dwayne, C.; Candace, C.-G.; Chirag, K.; Leah, N.; Elizabeth, P.; Umesh, H.; Alice, C.; Lisa, H.; Lois, L.-M.; et al. Modeling Liver Biology and the Tissue Response to Injury in Bioprinted Human Liver Tissues. Appl. Vitro Toxicol. 2018, 4, 288–303. [Google Scholar]
- Derakhshanfar, S.; Mbeleck, R.; Xu, K.; Zhang, X.; Zhong, W.; Xing, M. 3D bioprinting for biomedical devices and tissue engineering: A review of recent trends and advances. Bioact. Mater. 2018, 3, 144–156. [Google Scholar] [CrossRef]
- Yu, C.; Ma, X.; Zhu, W.; Wang, P.; Miller, K.L.; Stupin, J.; Koroleva-Maharajh, A.; Hairabedian, A.; Chen, S. Scanningless and continuous 3D bioprinting of human tissues with decellularized extracellular matrix. Biomaterials 2019, 194, 1–13. [Google Scholar] [CrossRef]
- Berg, J.; Hiller, T.; Kissner, M.S.; Qazi, T.H.; Duda, G.N.; Hocke, A.C.; Hippenstiel, S.; Elomaa, L.; Weinhart, M.; Fahrenson, C.; et al. Optimization of cell-laden bioinks for 3D bioprinting and efficient infection with influenza A virus. Sci. Rep. 2018, 8, 13877. [Google Scholar] [CrossRef]
- Lam, T.; Dehne, T.; Kruger, J.P.; Hondke, S.; Endres, M.; Thomas, A.; Lauster, R.; Sittinger, M.; Kloke, L. Photopolymerizable gelatin and hyaluronic acid for stereolithographic 3D bioprinting of tissue-engineered cartilage. J. Biomed. Mater. Res. B Appl. Biomater. 2019, 107, 2649–2657. [Google Scholar] [CrossRef]
- Van Den Bulcke, A.I.; Bogdanov, B.; De Rooze, N.; Schacht, E.H.; Cornelissen, M.; Berghmans, H. Structural and rheological properties of methacrylamide modified gelatin hydrogels. Biomacromolecules 2000, 1, 313–318. [Google Scholar] [CrossRef]
- Fairbanks, B.D.; Schwartz, M.P.; Bowman, C.N.; Anseth, K.S. Photoinitiated polymerization of PEG-diacrylate with lithium phenyl-2,4,6-trimethylbenzoylphosphinate: Polymerization rate and cytocompatibility. Biomaterials 2009, 30, 6702–6707. [Google Scholar] [CrossRef]
- Roth, S.J.; Hoper, D.; Beer, M.; Feineis, S.; Tischer, B.K.; Osterrieder, N. Recovery of infectious virus from full-length cowpox virus (CPXV) DNA cloned as a bacterial artificial chromosome (BAC). Vet. Res. 2011, 42, 3. [Google Scholar] [CrossRef]
- Witkowski, P.T.; Bourquain, D.; Bankov, K.; Auste, B.; Dabrowski, P.W.; Nitsche, A.; Kruger, D.H.; Schaade, L. Infection of human airway epithelial cells by different subtypes of Dobrava-Belgrade virus reveals gene expression patterns corresponding to their virulence potential. Virology 2016, 493, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Gelderblom, H.R.; Kocks, C.; L’Age-Stehr, J.; Reupke, H. Comparative immunoelectron microscopy with monoclonal antibodies on yellow fever virus-infected cells: Pre-embedding labelling versus immunocryoultramicrotomy. J. Virol. Methods 1985, 10, 225–239. [Google Scholar] [CrossRef]
- Uphoff, C.C.; Drexler, H.G. Detecting mycoplasma contamination in cell cultures by polymerase chain reaction. Methods Mol. Biol. 2011, 731, 93–103. [Google Scholar] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Kramski, M.; Meisel, H.; Klempa, B.; Kruger, D.H.; Pauli, G.; Nitsche, A. Detection and typing of human pathogenic hantaviruses by real-time reverse transcription-PCR and pyrosequencing. Clin. Chem. 2007, 53, 1899–1905. [Google Scholar] [CrossRef]
- Caliari, S.R.; Burdick, J.A. A practical guide to hydrogels for cell culture. Nat. Methods 2016, 13, 405–414. [Google Scholar] [CrossRef]
- Ades, E.W.; Candal, F.J.; Swerlick, R.A.; George, V.G.; Summers, S.; Bosse, D.C.; Lawley, T.J. HMEC-1: Establishment of an immortalized human microvascular endothelial cell line. J. Investig. Dermatol. 1992, 99, 683–690. [Google Scholar] [CrossRef]
- Abrams, G.A.; Murphy, C.J.; Wang, Z.Y.; Nealey, P.F.; Bjorling, D.E. Ultrastructural basement membrane topography of the bladder epithelium. Urol. Res. 2003, 31, 341–346. [Google Scholar]
- Abrams, G.A.; Schaus, S.S.; Goodman, S.L.; Nealey, P.F.; Murphy, C.J. Nanoscale topography of the corneal epithelial basement membrane and Descemet’s membrane of the human. Cornea 2000, 19, 57–64. [Google Scholar] [CrossRef]
- Baek, M.; Paick, S.H.; Jeong, S.J.; Hong, S.K.; Kim, S.W.; Choi, H. Urodynamic and histological changes in a sterile rabbit vesicoureteral reflux model. J. Korean Med. Sci. 2010, 25, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Walther, I.; Martinson, S.A.; Lopez, A.; Yason, C.; Godson, D.L.; Clark, E.G.; Simko, E. Porcine circovirus 2 inclusion bodies in pulmonary and renal epithelial cells. Vet. Pathol. 2008, 45, 640–644. [Google Scholar] [CrossRef] [PubMed]
- El-Hamamsy, I.; Chester, A.H.; Yacoub, M.H. Cellular regulation of the structure and function of aortic valves. J. Adv. Res. 2010, 1, 5–12. [Google Scholar] [CrossRef]
- Kurita, T.; Chitose, S.I.; Sato, K.; Sakazaki, T.; Fukahori, M.; Sueyoshi, S.; Umeno, H. Pathological mechanisms of laryngeal papillomatosis based on laryngeal epithelial characteristics. Laryngoscope Investig. Otolaryngol. 2019, 4, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Mine, S.; Fortunel, N.O.; Pageon, H.; Asselineau, D. Aging alters functionally human dermal papillary fibroblasts but not reticular fibroblasts: A new view of skin morphogenesis and aging. PLoS ONE 2008, 3, e4066. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D cell cultures-a comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Choi, D.J.; Park, S.J.; Gu, B.K.; Kim, Y.-J.; Chung, S.; Kim, C.-H. Effect of the pore size in a 3D bioprinted gelatin scaffold on fibroblast proliferation. J. Ind. Eng. Chem. 2018, 67, 388–395. [Google Scholar] [CrossRef]
- Lien, S.-M.; Ko, L.-Y.; Huang, T.-J. Effect of pore size on ECM secretion and cell growth in gelatin scaffold for articular cartilage tissue engineering. Acta Biomater. 2009, 5, 670–679. [Google Scholar] [CrossRef]
- Murphy, C.M.; Haugh, M.G.; O’Brien, F.J. The effect of mean pore size on cell attachment, proliferation and migration in collagen–glycosaminoglycan scaffolds for bone tissue engineering. Biomaterials 2010, 31, 461–466. [Google Scholar] [CrossRef]
- Jayadev, R.; Sherwood, D.R. Basement membranes. Curr. Biol. CB 2017, 27, R207–R211. [Google Scholar] [CrossRef]
- O’Brien, F.J.; Harley, B.A.; Yannas, I.V.; Gibson, L.J. The effect of pore size on cell adhesion in collagen-GAG scaffolds. Biomaterials 2005, 26, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Yannas, I.V.; Lee, E.; Orgill, D.P.; Skrabut, E.M.; Murphy, G.F. Synthesis and characterization of a model extracellular matrix that induces partial regeneration of adult mammalian skin. Proc. Natl. Acad. Sci. USA 1989, 86, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Haycock, J.W. 3D cell culture: A review of current approaches and techniques. Methods Mol. Biol. 2011, 695, 1–15. [Google Scholar] [PubMed]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Vinci, M.; Gowan, S.; Boxall, F.; Patterson, L.; Zimmermann, M.; Court, W.; Lomas, C.; Mendiola, M.; Hardisson, D.; Eccles, S.A. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012, 10, 29. [Google Scholar] [CrossRef]
- Graf, B.W.; Boppart, S.A. Imaging and analysis of three-dimensional cell culture models. Methods Mol. Biol. 2010, 591, 211–227. [Google Scholar]
- Hudu, S.A.; Alshrari, A.S.; Syahida, A.; Sekawi, Z. Cell Culture, Technology: Enhancing the Culture of Diagnosing Human Diseases. J. Clin. Diagn. Res. 2016, 10, DE01–DE05. [Google Scholar] [CrossRef]
- Leland, D.S.; Ginocchio, C.C. Role of cell culture for virus detection in the age of technology. Clin. Microbiol. Rev. 2007, 20, 49–78. [Google Scholar] [CrossRef]
- Berto, A.; Van der Poel, W.H.; Hakze-van der Honing, R.; Martelli, F.; La Ragione, R.M.; Inglese, N.; Collins, J.; Grierson, S.; Johne, R.; Reetz, J.; et al. Replication of hepatitis E virus in three-dimensional cell culture. J. Virol. Methods 2013, 187, 327–332. [Google Scholar] [CrossRef]
- Pauli, G.; Blümel, J.; Burger, R.; Drosten, C.; Gröner, A.; Gürtler, L.; Heiden, M.; Hildebrandt, M.; Jansen, B.; Montag-Lessing, T.; et al. Orthopox Viruses: Infections in Humans. Transfus. Med. Hemother 2010, 37, 351–364. [Google Scholar]
- Verheust, C.; Goossens, M.; Pauwels, K.; Breyer, D. Biosafety aspects of modified vaccinia virus Ankara (MVA)-based vectors used for gene therapy or vaccination. Vaccine 2012, 30, 2623–2632. [Google Scholar] [CrossRef] [PubMed]
- Yakes, B.J.; Papafragkou, E.; Conrad, S.M.; Neill, J.D.; Ridpath, J.F.; Burkhardt, W., III; Kulka, M.; Degrasse, S.L. Surface plasmon resonance biosensor for detection of feline calicivirus, a surrogate for norovirus. Int. J. Food Microbiol. 2013, 162, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Sironen, T.; Vaheri, A.; Plyusnin, A. Molecular evolution of Puumala hantavirus. J. Virol. 2001, 75, 11803–11810. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.D.; Barrett, A.D.T. Live Attenuated Yellow Fever 17D Vaccine: A Legacy Vaccine Still Controlling Outbreaks In Modern Day. Curr. Infect. Dis. Rep. 2017, 19, 14. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.F.; Yellayi, S.; Cann, J.A.; Johnson, A.; Smith, A.L.; Paragas, J.; Jahrling, P.B.; Blaney, J.E. Cowpox virus infection of cynomolgus macaques as a model of hemorrhagic smallpox. Virology 2011, 418, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Cromeans, T.; Park, G.W.; Costantini, V.; Lee, D.; Wang, Q.; Farkas, T.; Lee, A.; Vinje, J. Comprehensive comparison of cultivable norovirus surrogates in response to different inactivation and disinfection treatments. Appl. Environ. Microbiol. 2014, 80, 5743–5751. [Google Scholar] [CrossRef]
- Rissanen, I.; Stass, R.; Zeltina, A.; Li, S.; Hepojoki, J.; Harlos, K.; Gilbert, R.J.C.; Huiskonen, J.T.; Bowden, T.A. Structural Transitions of the Conserved and Metastable Hantaviral Glycoprotein Envelope. J. Virol. 2017, 91, 21. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koban, R.; Lam, T.; Schwarz, F.; Kloke, L.; Bürge, S.; Ellerbrok, H.; Neumann, M. Simplified Bioprinting-Based 3D Cell Culture Infection Models for Virus Detection. Viruses 2020, 12, 1298. https://doi.org/10.3390/v12111298
Koban R, Lam T, Schwarz F, Kloke L, Bürge S, Ellerbrok H, Neumann M. Simplified Bioprinting-Based 3D Cell Culture Infection Models for Virus Detection. Viruses. 2020; 12(11):1298. https://doi.org/10.3390/v12111298
Chicago/Turabian StyleKoban, Robert, Tobias Lam, Franziska Schwarz, Lutz Kloke, Silvio Bürge, Heinz Ellerbrok, and Markus Neumann. 2020. "Simplified Bioprinting-Based 3D Cell Culture Infection Models for Virus Detection" Viruses 12, no. 11: 1298. https://doi.org/10.3390/v12111298
APA StyleKoban, R., Lam, T., Schwarz, F., Kloke, L., Bürge, S., Ellerbrok, H., & Neumann, M. (2020). Simplified Bioprinting-Based 3D Cell Culture Infection Models for Virus Detection. Viruses, 12(11), 1298. https://doi.org/10.3390/v12111298