Polymorphisms in Processing and Antigen Presentation-Related Genes and Their Association with Host Susceptibility to Influenza A/H1N1 2009 Pandemic in a Mexican Mestizo Population

 ,
,  ,
,  ,
, 
Abstract

1. Introduction
2. Materials and Methods
2.1. Ethics Statements
2.2. Diagnosis
2.3. Population
2.4. DNA Extraction
2.5. SNP Selection
2.6. SNP Genotyping
2.7. Bioinformatic Analysis and in Silico Analysis
2.8. Hardy–Weinberg Equilibrium and Haplotypes
2.9. Statistical Analysis
3. Results
3.1. Demographic Variables in Case and Control Groups
3.2. Demographic and Clinical Variables in Survivors and Nonsurvivors
3.3. Hardy–Weinberg Equilibrium
3.4. Allele and Genotype Frequencies
3.5. Allele and Genotype Frequencies from Survivors and Nonsurvivors
3.6. Logistic Regression Analysis
3.7. Logistic Regression Analysis in Survivors and Nonsurvivors
3.8. Bioinformatic Analysis
3.9. In Silico Analysis
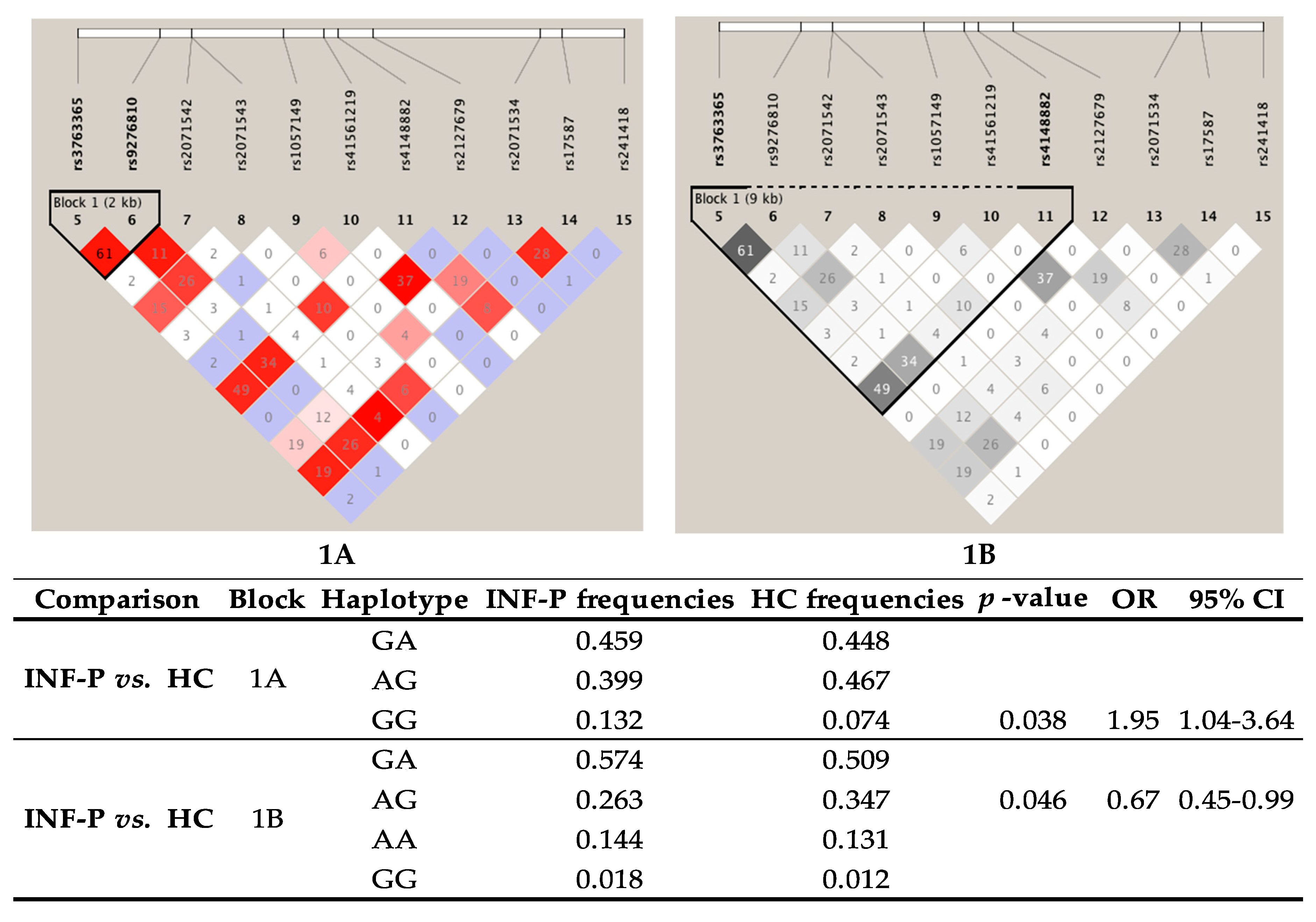
3.10. Linkage Disequilibrium (LD) and Haplotype Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Variables | Survivors = 109 | Non-Survivors = 19 | p-Value |
|---|---|---|---|
| Age | 39 (20–89) | 44 (30–56) | 0.93 |
| Sex, male (%) | 64 (58.72) | 13 (68.42) | 0.43 |
| BMI | 30.9 (17.4–52.7) | 32.2 (24.4–44.6) | 0.99 |
| Tobacco smoking (%) | 49 (44.95) | 8 (42.11) | 0.82 |
| Comorbidities | |||
| Hypertension (%) | 17 (15.6) | 0 | 0.07 |
| Asthma (%) | 9 (8.26) | 1 (5.26) | 0.65 |
| Diabetes (%) | 8 (7.34) | 2 (10.53) | 0.64 |
| COPD (%) | 4 (3.67) | 0 | 1 |
| Symptomatology | |||
| Fever (%) | 85 (77.68) | 12 (63.16) | 0.16 |
| Dyspnea (%) | 79 (72.48) | 14 (73.68) | 1 |
| Cough (%) | 58 (53.21) | 10 (52.63) | 0.96 |
| Rhinorrhea (%) | 38 (34.86) | 4 (21.05) | 0.29 |
| Nasal congestion (%) | 15 (13.76) | 0 | 0.12 |
| Clinical variables | |||
| Leukocytes | 9.5 (2.7–15) | 10.7 (1.9–10.9) | 0.87 |
| Platelets | 208 (261–551) | 153 (62–233) | 0.006 |
| Glucose | 107 (43–341) | 122 (113–189) | 0.007 |
| Urea | 26.5 (8–178) | 38 (5–63) | 0.89 |
| Creatinine | 0.8 (0.25–10.1) | 0.9 (0.75–1.35) | 0.25 |
| CPK | 226 (11–1438) | 670 (20–7241) | 0.012 |
| LDH | 481 (104–1481) | 1082 (524–1477) | <0.001 |
| AST | 45 (16–184) | 69 (15–252) | 0.22 |
| ALT | 38 (10–174) | 37 (12–124) | 0.63 |
| Complications | |||
| Pneumonia (%) | 67 (61.47) | 9 (47.37) | 0.25 |
| ICU (%) | 32 (29.35) | 14 (73.68) | <0.001 |
| ARDS (%) | 29 (26.61) | 9 (47.37) | 0.06 |
| Gene | Non-Survivors | Survivors | p-Value | OR | 95% CI | ||
|---|---|---|---|---|---|---|---|
| n | F (%) | n | F (%) | ||||
| TAP2 | rs241433 | ||||||
| Genotypes | |||||||
| AA | 11 | 57.89 | 64 | 58.72 | 0.95 | 0.97 | 0.36–2.60 |
| AC | 8 | 42.11 | 45 | 41.28 | 1.03 | 0.39–2.78 | |
| CC | 0 | 0 | 0 | 0 | |||
| 19 | 100 | 109 | 100 | ||||
| Alleles | |||||||
| A | 30 | 79 | 173 | 79.36 | 0.95 | 0.98 | 0.42–2.27 |
| C | 8 | 21 | 45 | 20.64 | 1.03 | 0.44–2.39 | |
| TAPBP | rs2071888 | ||||||
| Genotypes | |||||||
| CC | 6 | 31.58 | 18 | 16.67 | 0.11 | 1 | |
| CG | 7 | 36.84 | 67 | 62.04 | 0.31 | 0.09–1.05 | |
| GG | 6 | 31.58 | 23 | 21.30 | 1.28 | 0.35–4.64 | |
| 19 | 100 | 108 | 100 | ||||
| Alleles | |||||||
| C | 19 | 50 | 103 | 48 | 0.79 | 1.09 | 0.55–2.19 |
| G | 19 | 50 | 113 | 52 | 0.91 | 0.46–1.82 | |
| TAPBP | rs2282851 | ||||||
| Genotypes | |||||||
| CC | 7 | 36.84 | 29 | 26.85 | 0.130 | 1 | |
| CT | 7 | 36.84 | 65 | 60.19 | 0.45 | 0.14–1.38 | |
| TT | 5 | 26.32 | 14 | 12.96 | 1.48 | 0.40–5.50 | |
| 19 | 100 | 108 | 100 | ||||
| Alleles | |||||||
| C | 21 | 55 | 123 | 56.94 | 0.85 | 0.93 | 0.47–1.87 |
| T | 17 | 45 | 93 | 43.06 | 1.07 | 0.53–2.14 | |
Appendix B

References
- Perez-Padilla, R.; De La Rosa-Zamboni, D.; De Leon, S.P.; Hernandez, M.; Quiñones-Falconi, F.; Bautista, E.; Ramirez-Venegas, A.; Rojas-Serrano, J.; Ormsby, C.E.; Corrales, A.; et al. Pneumonia and Respiratory Failure from Swine-Origin Influenza A (H1N1) in Mexico. N. Engl. J. Med. 2009, 361, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Echevarría-Zuno, S.; Mejía-Aranguré, J.M.; Mar-Obeso, A.J.; Grajales-Muñiz, C.; Robles-Pérez, E.; González-León, M.; Ortega-Alvarez, M.C.; Gonzalez-Bonilla, C.; Rascón-Pacheco, R.A.; Borja-Aburto, V.H. Infection and death from influenza A H1N1 virus in Mexico: A retrospective analysis. Lancet 2009, 374, 2072–2079. [Google Scholar] [CrossRef]
- Ward, K.A.; Spokes, P.J.; McAnulty, J.M. Case-control Study of Risk Factors for Hospitalization Caused by Pandemic (H1N1) 2009. Emerg. Infect. Dis. 2011, 17, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Fezeu, L.; Julia, C.; Henegar, A.; Bitu, J.; Hu, F.B.; Grobbee, D.E.; Kengne, A.P.; Hercberg, S.; Czernichow, S. Obesity is associated with higher risk of intensive care unit admission and death in influenza A (H1N1) patients: A systematic review and meta-analysis. Obes. Rev. 2011, 12, 653–659. Available online: https://pubmed.ncbi.nlm.nih.gov/21457180/ (accessed on 12 July 2020). [CrossRef] [PubMed]
- Chowell, G.; Echevarría-Zuno, S.; Viboud, C.; Simonsen, L.; Tamerius, J.; Miller, M.A.; Borja-Aburto, V.H. Characterizing the Epidemiology of the 2009 Influenza A/H1N1 Pandemic in Mexico. PLoS Med. 2011, 8, e1000436. [Google Scholar] [CrossRef] [PubMed]
- Carlos, J.; Salvador, N.; Marcela, L.; Garduño, B.; Molina, H.L.; Serrano, A.T. Factores predictores de defunción en pacientes con neumonía por influenza a H1N1. Med. Interna México 2011, 27. [Google Scholar]
- Shi, S.J.; Li, H.; Liu, M.; Liu, Y.M.; Zhou, F.; Liu, B.; Qu, J.X.; Cao, B. Mortality prediction to hospitalized patients with influenza pneumonia: PO2/FiO2combined lymphocyte count is the answer. Clin. Respir. J. 2015, 11, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Mata-Marín, L.A.; Mata-Marín, J.A.; Vásquez-Mota, V.C.; Arroyo-Anduiza, C.I.; Gaytán-Martínez, J.; Manjarrez-Téllez, B.; Ochoa-Carrera, L.A.; Sandoval-Ramirez, J.L. Risk factors associated with mortality in patients infected with influenza A/H1N1 in Mexico. BMC Res. Notes 2015, 8, 432. [Google Scholar] [CrossRef][Green Version]
- Bermejo-Martin, J.F.; De Lejarazu, R.O.; Pumarola, T.; Rello, J.; Almansa, R.; Ramírez, P.; Martin-Loeches, I.; Varillas, D.; Gallegos, M.C.; Serón, C.; et al. Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza. Crit. Care 2009, 13, R201. [Google Scholar] [CrossRef]
- To, K.K.W.; Hung, I.F.N.; Li, I.W.S.; Lee, K.; Koo, C.; Yan, W.; Liu, R.; Ho, K.; Chu, K.; Watt, C.; et al. Delayed Clearance of Viral Load and Marked Cytokine Activation in Severe Cases of Pandemic H1N1 2009 Influenza Virus Infection. Clin. Infect. Dis. 2010, 50, 850–859. [Google Scholar] [CrossRef]
- Garcia-Ramirez, R.A.; Ramirez-Venegas, A.; Quintana-Carrillo, R.; Camarena, Á.E.; Falfán-Valencia, R.; Mejía-Aranguré, J.M. TNF, IL6, and IL1B Polymorphisms Are Associated with Severe Influenza A (H1N1) Virus Infection in the Mexican Population. PLOS ONE 2015, 10, e0144832. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Venegas, A.; Gonz lez-Bonilla, C.; Borja-Aburto, V. Pandemic influenza A/H1N1 virus infection and TNF, LTA, IL1B, IL6, IL8, and CCL polymorphisms in Mexican population: A case-control study. BMC Infect. Dis. 2012, 12, 299–307. Available online: http://www.biomedcentral.com/1471-2334/12/299 (accessed on 28 July 2020).
- Yewdell, J.W.; Bennink, J.R.; Hosaka, Y. Cells process exogenous proteins for recognition by cytotoxic T lymphocytes. Science 1988, 239, 637–640. [Google Scholar] [CrossRef]
- Wahl, A.; Schafer, F.; Bardet, W.; Buchli, R.; Air, G.M.; Hildebrand, W.H. HLA class I molecules consistently present internal influenza epitopes. Proc. Natl. Acad. Sci. USA 2009, 106, 540–545. Available online: www.pnas.org/cgi/content/full/ (accessed on 28 July 2020). [CrossRef]
- Ortmann, B.; Copeman, J.; Lehner, P.J.; Sadasivan, B.; Herberg, J.A.; Grandea, A.G.; Riddell, S.R.; Tampé, R.; Spies, T.; Trowsdale, J.; et al. A Critical Role for Tapasin in the Assembly and Function of Multimeric MHC Class I-TAP Complexes. Science 1997, 277, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sjögren, H.-O.; Hellman, U.; Pettersson, R.F.; Wang, P. Cloning and functional characterization of a subunit of the transporter associated with antigen processing. Proc. Natl. Acad. Sci. USA 1997, 94, 8708–8713. Available online: www.pnas.org (accessed on 28 July 2020). [CrossRef]
- Rizvi, S.M.; Raghavan, M. Mechanisms of function of tapasin, a critical major histocompatibility complex class I assembly factor. Traffic 2009, 11, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Momburgu, F.; Liuu, T.; Motalu, U.M.A.; Jondalu, M.; Hämmerlingu, G.J.; Ljunggren, H.-G.; Momburg, F.; Liu, T.; Motal, U.M.A.; et al. Presentation of viral antigens restricted by H-2Kb, Db or Kd in proteasome subunit LMP2-and LMP7-deficient cells. Eur. J. Immunol. 1994, 24, 1863–1868. [Google Scholar] [CrossRef]
- Falfán-Valencia, R.; Narayanankutty, A.; Reséndiz-Hernández, J.M.; Pérez-Rubio, G.; Ramírez-Venegas, A.; Nava-Quiroz, K.J.; Bautista-Félix, N.E.; Vargas-Alarcón, G.; Castillejos-López, M.D.J.; Hernández, A. An Increased Frequency in HLA Class I Alleles and Haplotypes Suggests Genetic Susceptibility to Influenza A (H1N1) 2009 Pandemic: A Case-Control Study. J. Immunol. Res. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dutta, M.; Dutta, P.; Medhi, S.; Borkakoty, B.; Biswas, D. Polymorphism of HLA class I and class II alleles in influenza A(H1N1)pdm09 virus infected population of Assam, Northeast India. J. Med Virol. 2018, 90, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Camarena, A.; Aquino-Gálvez, A.; Falfán-Valencia, R.; Sanchez, G.; Montaño, M.; Ramos, C.; Juárez, A.; De Alba, C.G.; Granados, J.; Selman, M. PSMB8 (LMP7) but not PSMB9 (LMP2) gene polymorphisms are associated to pigeon breeder’s hypersensitivity pneumonitis. Respir. Med. 2010, 104, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Yin, B.; Shen, T.; Ma, Q.; Liu, L.; Zheng, J.; Zhao, Y.; Qian, K.; Liu, D. TAP1 and TAP2 polymorphisms associated with ankylosing spondylitis in genetically homogenous Chinese Han population. Hum. Immunol. 2009, 70, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.M.; Jordanova, E.S.; Corver, W.E.; Ven Wezel, T.; Uh, H.W.; Kenter, G.G. Single nucleotide polymorphisms in antigen processing machinery component ERAPI significantly associate with clinical outcome in cervical carcinoma. Genes Chromosomes Cancer 2009, 48, 410–418. Available online: https://pubmed.ncbi.nlm.nih.gov/19202550/ (accessed on 28 July 2020). [CrossRef] [PubMed]
- Deshpande, A.; Wheeler, C.M.; Hunt, W.C.; Peyton, C.L.; White, P.S.; Valdez, Y.E.; Nolan, J.P. Variation in HLA Class I Antigen-Processing Genes and Susceptibility to Human Papillomavirus Type 16–Associated Cervical Cancer. J. Infect. Dis. 2008, 197, 371–381. [Google Scholar] [CrossRef]
- Little, J.; Higgins, J.P.T.; Ioannidis, J.P.; Moher, D.; Gagnon, F.; Von Elm, E.; Khoury, M.J.; Cohen, B.; Davey-Smith, G.; Grimshaw, J.; et al. STrengthening the REporting of Genetic Association studies (STREGA)—An extension of the STROBE statement. Eur. J. Clin. Investig. 2009, 39, 247–266. [Google Scholar] [CrossRef]
- Ensembl Genome Browser 89. Available online: http://www.ensembl.org/index.html (accessed on 4 August 2017).
- Ward, L.D.; Kellis, M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012, 40, D930–D934. [Google Scholar] [CrossRef]
- SNPStats: A Web Tool for the Analysis of Association Studies—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/16720584/ (accessed on 23 June 2020).
- Haploview: Analysis and Visualization of LD and Haplotype Maps—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/15297300/ (accessed on 23 June 2020).
- Gabriel, S.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The Structure of Haplotype Blocks in the Human Genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef]
- Dean, A.G.; Arner, T.G.; Sunki, G.G.; Friedman, R.; Lantinga, M.; Sangam, S.; Zubieta, J.C.; Sullivan, K.M.; Brendel, K.A.; Gao, Z.; et al. Epi Info, A Database and Statistics Program for Public Health Professionals; CDC: Atlanta, GA, USA, 2004.
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Lenzi, L.; De Mello, Â.M.; Da Silva, L.R.; Grochocki, M.H.C.; Pontarolo, R. Influenza pandêmica A (H1N1) 2009: fatores de risco para o internamento. J. Bras. Pneumol. 2012, 38, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Ambrocio-Ortiz, E.; Pérez-Rubio, G.; Ramírez-Venegas, A.; Hernández-Zenteno, R.; Del Angel-Pablo, A.D.; Pérez-Rodríguez, M.E.; Salazar, A.M.; Abarca-Rojano, E.; Falfán-Valencia, R. Effect of SNPs in HSP Family Genes, Variation in the mRNA and Intracellular Hsp Levels in COPD Secondary to Tobacco Smoking and Biomass-Burning Smoke. Front. Genet. 2020, 10, 1307. [Google Scholar] [CrossRef] [PubMed]
- Namipashaki, A.; Razaghi-Moghadam, Z.; Ansari-Pour, N. The essentiality of reporting Hardy-Weinberg equilibrium calculations in population-based genetic association studies. Cell J. Royan Inst. 2015, 17, 187–192. [Google Scholar]
- Braud, V.M.; McMichael, A.J.; Cerundolo, V. Differential processing of influenza nucleoprotein in human and mouse cells. Eur. J. Immunol. 1998, 28, 625–635. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/%28SICI%291521-4141%28199802%2928%3A02%3C625%3A%3AAID-IMMU625%3E3.0.CO%3B2-I (accessed on 29 July 2020). [CrossRef]
- Ho, A.W.S.; Prabhu, N.; Betts, R.J.; Ge, M.Q.; Dai, X.; Hutchinson, P.E.; Lew, F.C.; Wong, K.L.; Hanson, B.J.; Macary, P.A.; et al. Lung CD103 + Dendritic Cells Efficiently Transport Influenza Virus to the Lymph Node and Load Viral Antigen onto MHC Class I for Presentation to CD8 T Cells. J. Immunol. 2011, 187, 6011–6021. [Google Scholar] [CrossRef]
- Mehta, A.M.; Jordanova, E.S.; Van Wezel, T.; Uh, H.-W.; Corver, W.E.; Kwappenberg, K.M.C.; Verduijn, W.; Kenter, G.G.; Van Der Burg, S.H.; Fleuren, G.J. Genetic variation of antigen processing machinery components and association with cervical carcinoma. Genes Chromosomes Cancer 2007, 46, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-S.; Lee, A.T.; Criswell, L.A.; Seldin, M.F.; Amos, C.I.; Carulli, J.P.; Navarrete, C.; Remmers, E.F.; Kastner, D.L.; Plenge, R.M.; et al. Several Regions in the Major Histocompatibility Complex Confer Risk for Anti-CCP-Antibody Positive Rheumatoid Arthritis, Independent of the DRB1 Locus. Mol. Med. 2008, 14, 293–300. [Google Scholar] [CrossRef]
- HNRNPA1 Heterogeneous Nuclear Ribonucleoprotein A1 [Homo Sapiens (Human)]—Gene—NCBI. [cited]. Available online: https://www.ncbi.nlm.nih.gov/gene/3178 (accessed on 9 September 2020).
- Kelly, A.; Trowsdale, J. Genetics of antigen processing and presentation. Immunogenetics 2018, 71, 161–170. [Google Scholar] [CrossRef]
- Harvey, I.B.; Wang, X.; Fremont, D.H. Molluscum contagiosum virus MC80 sabotages MHC-I antigen presentation by targeting tapasin for ER-associated degradation. PLoS Pathog. 2019, 15, e1007711. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Zhang, Y.; Lu, X.; Xu, Y.; Wang, J.; Zhang, Y.; Yu, R.; Su, J. Association of Polymorphisms in HLA Antigen Presentation-Related Genes with the Outcomes of HCV Infection. PLoS ONE 2015, 10, e0123513. [Google Scholar] [CrossRef][Green Version]
- O’Brien, K.M.; Orlow, I.; Antonescu, C.R.; Ballman, K.V.; McCall, L.; DeMatteo, R.P.; Engel, L.S. Gastrointestinal stromal tumors: A case-only analysis of single nucleotide polymorphisms and somatic mutations. Clin. Sarcoma Res. 2013, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-H.; Park, J.-S.; Park, B.L.; Bae, D.-J.; Uh, S.-T.; Kim, M.-K.; Choi, I.S.; Shin, H.D.; Park, C.-S. Association analysis of tapasin polymorphisms with aspirin-exacerbated respiratory disease in asthmatics. Pharm. Genom. 2013, 23, 341–348. [Google Scholar] [CrossRef]
- TAPBP—Tapasin Precursor—Homo Sapiens (Human)—TAPBP Gene & Protein. Available online: https://www.uniprot.org/uniprot/O15533 (accessed on 9 September 2020).
- UniProtKB/SwissProt Variant VAR_010253. Available online: https://web.expasy.org/variant_pages/VAR_010253.html (accessed on 9 September 2020).
- Chen, T.; Xiao, M.-F.; Yang, J.; Chen, Y.K.; Bai, T.; Tang, X.J.; Shu, Y. Association between rs12252 and influenza susceptibility and severity: An updated meta-analysis. Epidemiol. Infect. 2018, 147, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Glynne, R.; Powis, S.H.; Beck, S.; Kelly, A.; Kerr, L.-A.; Trowsdale, J. A proteasome-related gene between the two ABC transporter loci in the class II region of the human MHC. Nature 1991, 353, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Zúñiga, J.; Buendia-Roldan, I.; Zhao, Y.; Jiménez, L.; Torres, D.; Romo, J.; Ramirez, G.; Cruz, A.; Vargas-Alarcón, G.; Sheu, C.-C.; et al. Genetic variants associated with severe pneumonia in A/H1N1 influenza infection. Eur. Respir. J. 2011, 39, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Chatzopoulou, F.; Gioula, G.; Kioumis, I.; Chatzidimitriou, D.; Exindari, M. Identification of complement-related host genetic risk factors associated with influenza A(H1N1)pdm09 outcome: Challenges ahead. Med. Microbiol. Immunol. 2019, 208, 631–640. [Google Scholar] [CrossRef]

| Variables | INF-P = 128 | HC = 111 | p-Value |
|---|---|---|---|
| Age | 41 (19–86) | 44 (19–82) | 0.986 |
| Sex, male (%) | 77 (60.16) | 46 (41.44) | 0.003 |
| BMI | 30.5 (17.4–52.8) | 26.8 (16–44.8) | <0.001 |
| Tobacco smoking, % | 57 (44.53) | 39 (35.14) | 0.139 |
| Gene | INF-P | HC | p-Value | p-Value Corrected | OR | 95% CI | ||
|---|---|---|---|---|---|---|---|---|
| n | F (%) | n | F (%) | |||||
| TAP2 | rs241433 | |||||||
| Genotypes | ||||||||
| AA | 75 | 58.59 | 28 | 25.23 | <0.0001 | <0.0001 | 4.19 | 2.41–7.30 |
| AC | 53 | 41.41 | 83 | 74.77 | 0.24 | 0.14–0.42 | ||
| CC | 0 | 0 | 0 | 0 | ||||
| 128 | 100 | 111 | 100 | |||||
| Alleles | ||||||||
| A | 203 | 79 | 139 | 62.61 | <0.0001 | <0.0001 | 2.29 | 1.52–3.44 |
| C | 53 | 21 | 83 | 37.39 | 0.44 | 0.29–0.66 | ||
| TAPBP | rs2071888 | |||||||
| Genotypes | ||||||||
| CC | 24 | 18.90 | 28 | 25.45 | 0.0008 | 0.014 | 1 | |
| CG | 74 | 58.27 | 76 | 69.09 | 1.14 | 0.60–2.14 | ||
| GG | 29 | 22.83 | 6 | 5.45 | 5.64 | 2.00–15.86 | ||
| 127 | 100 | 110 | 100 | |||||
| Alleles | ||||||||
| C | 122 | 48.03 | 132 | 60 | 0.009 | 0.153 | 0.62 | 0.43–0.89 |
| G | 132 | 51.97 | 88 | 40 | 1.62 | 1.13–2.34 | ||
| TAPBP | rs2282851 | |||||||
| Genotypes | ||||||||
| CC | 36 | 29.03 | 15 | 13.64 | 0.017 | 0.289 | 1 | |
| CT | 69 | 55.65 | 75 | 68.18 | 0.38 | 0.19–0.76 | ||
| TT | 19 | 15.32 | 20 | 18.18 | 0.40 | 0.17–0.94 | ||
| 124 | 100 | 110 | 100 | |||||
| Alleles | ||||||||
| C | 141 | 57 | 105 | 47.73 | 0.048 | 0.816 | 1.44 | 1.00–2.08 |
| T | 107 | 43 | 115 | 52.27 | 0.69 | 0.40–0.99 | ||
| Alleles | |||||||
|---|---|---|---|---|---|---|---|
| Chr | Gene | SNP | A1 | Test | p-Value | OR | 95% CI |
| 6 | TAP2 | rs241433 | C | Add | <0.0001 | 0.241 | 0.133–0.434 |
| Sex | 0.03979 | 1.828 | 1.029–3.247 | ||||
| BMI | <0.0001 | 1.102 | 1.05–1.157 | ||||
| TAPBP | rs2071888 | G | Add | 0.0095 | 1.891 | 1.168–3.061 | |
| Sex | 0.0056 | 2.184 | 1.257–3.794 | ||||
| BMI | 0.0001 | 1.097 | 1.046–1.15 | ||||
| Genotypes | |||||||
| Chr | Gene | SNP | A1 | Test | p-Value | OR | 95% CI |
| 6 | TAP2 | rs241433 | CC | Add | NA | NA | NA |
| Domdev | NA | NA | NA | ||||
| Sex | NA | NA | NA | ||||
| BMI | NA | NA | NA | ||||
| Geno_2DF | NA | NA | NA | ||||
| TAPBP | rs2071888 | GG | Add | 0.005 | 2.184 | 1.274–3.745 | |
| Domdev | 0.075 | 0.5612 | 0.297–1.059 | ||||
| Sex | 0.009 | 2.102 | 1.205–3.668 | ||||
| BMI | 0.0002 | 1.093 | 1.043–1.145 | ||||
| Geno_2DF | 0.013 | NA | NA | ||||
| SNP Allele | Sequence | Absolute Affinity Value | Protein |
|---|---|---|---|
| rs241433/C | GGATATACAGTCCCTTCTCCTACCATACAG | 12.4 | UP1 |
| rs241433/A | GGATATACAGTCCATTCTCCTACCATACAG | 0.4 | |
| rs2071888/G | TTGAACTGTAGGCAGCC | 0.8 | HNF4 |
| rs2071888/C | TTGAACTCTAGGCAGCC | 10.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponce-Gallegos, M.A.; Ruiz-Celis, A.; Ambrocio-Ortiz, E.; Pérez-Rubio, G.; Ramírez-Venegas, A.; Bautista-Félix, N.E.; Falfán-Valencia, R. Polymorphisms in Processing and Antigen Presentation-Related Genes and Their Association with Host Susceptibility to Influenza A/H1N1 2009 Pandemic in a Mexican Mestizo Population. Viruses 2020, 12, 1224. https://doi.org/10.3390/v12111224
Ponce-Gallegos MA, Ruiz-Celis A, Ambrocio-Ortiz E, Pérez-Rubio G, Ramírez-Venegas A, Bautista-Félix NE, Falfán-Valencia R. Polymorphisms in Processing and Antigen Presentation-Related Genes and Their Association with Host Susceptibility to Influenza A/H1N1 2009 Pandemic in a Mexican Mestizo Population. Viruses. 2020; 12(11):1224. https://doi.org/10.3390/v12111224
Chicago/Turabian StylePonce-Gallegos, Marco Antonio, Aseneth Ruiz-Celis, Enrique Ambrocio-Ortiz, Gloria Pérez-Rubio, Alejandra Ramírez-Venegas, Nora E. Bautista-Félix, and Ramcés Falfán-Valencia. 2020. "Polymorphisms in Processing and Antigen Presentation-Related Genes and Their Association with Host Susceptibility to Influenza A/H1N1 2009 Pandemic in a Mexican Mestizo Population" Viruses 12, no. 11: 1224. https://doi.org/10.3390/v12111224
APA StylePonce-Gallegos, M. A., Ruiz-Celis, A., Ambrocio-Ortiz, E., Pérez-Rubio, G., Ramírez-Venegas, A., Bautista-Félix, N. E., & Falfán-Valencia, R. (2020). Polymorphisms in Processing and Antigen Presentation-Related Genes and Their Association with Host Susceptibility to Influenza A/H1N1 2009 Pandemic in a Mexican Mestizo Population. Viruses, 12(11), 1224. https://doi.org/10.3390/v12111224