Maturation of Pseudo-Nucleus Compartment in P. aeruginosa, Infected with Giant phiKZ Phage


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacteriophage, Bacterial Strain and Growth Conditions
2.2. DNA Extraction, Agarose Electrophoresis, and Real-Time PCR
2.3. Fluorescent In Situ Hybridization
2.4. Transmission Electron Microscopy
2.5. Sample Preparation for Analytical Electron Microscopy and Electron Tomography
2.6. Electron Tomography
2.7. Energy Dispersive X-ray Spectroscopy (EDX)
2.8. Electron Energy Loss Spectroscopy (EELS)
3. Results
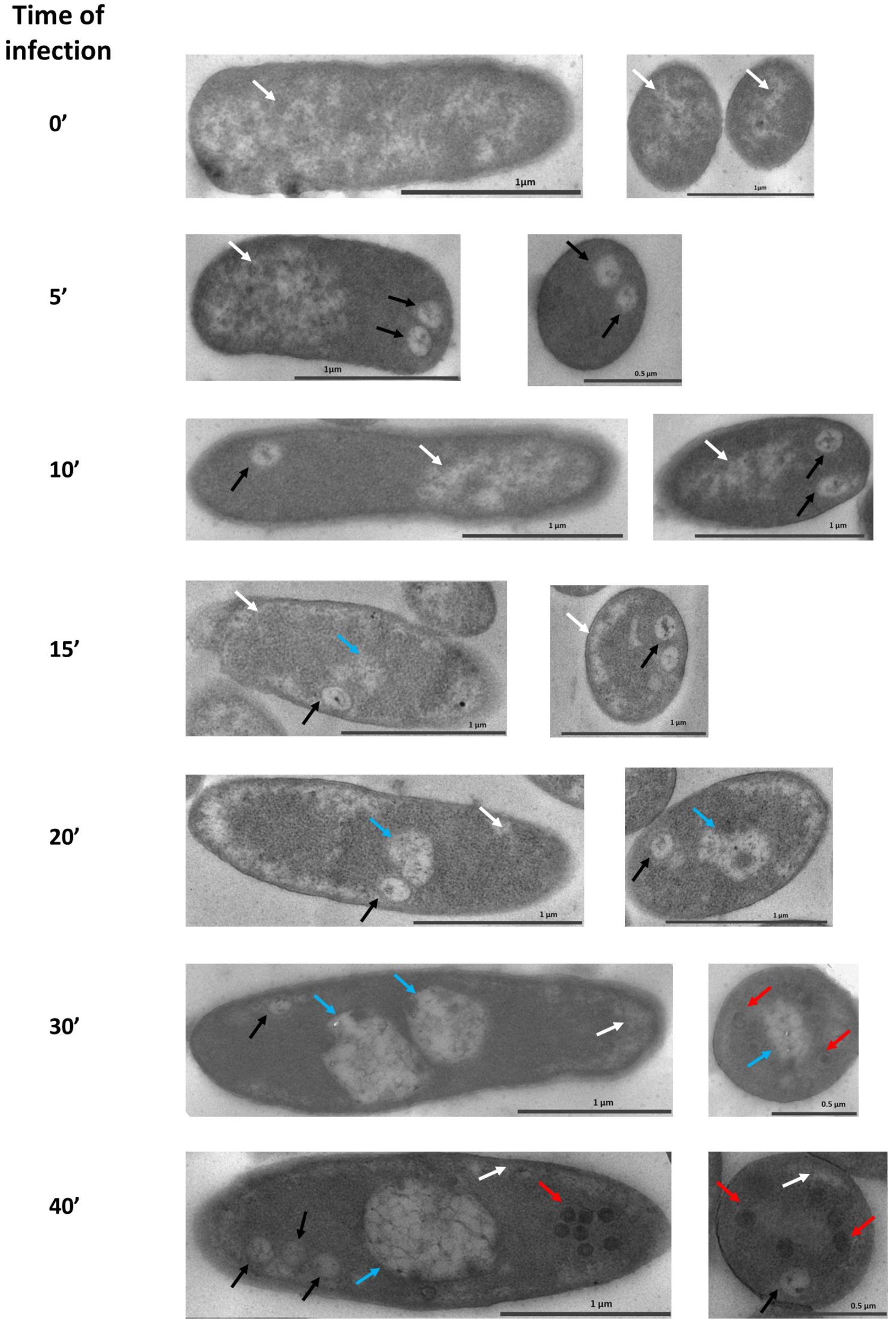
3.1. Maturation of the Pseudo-Nucleus in P. aeruginosa after phiKZ Infection
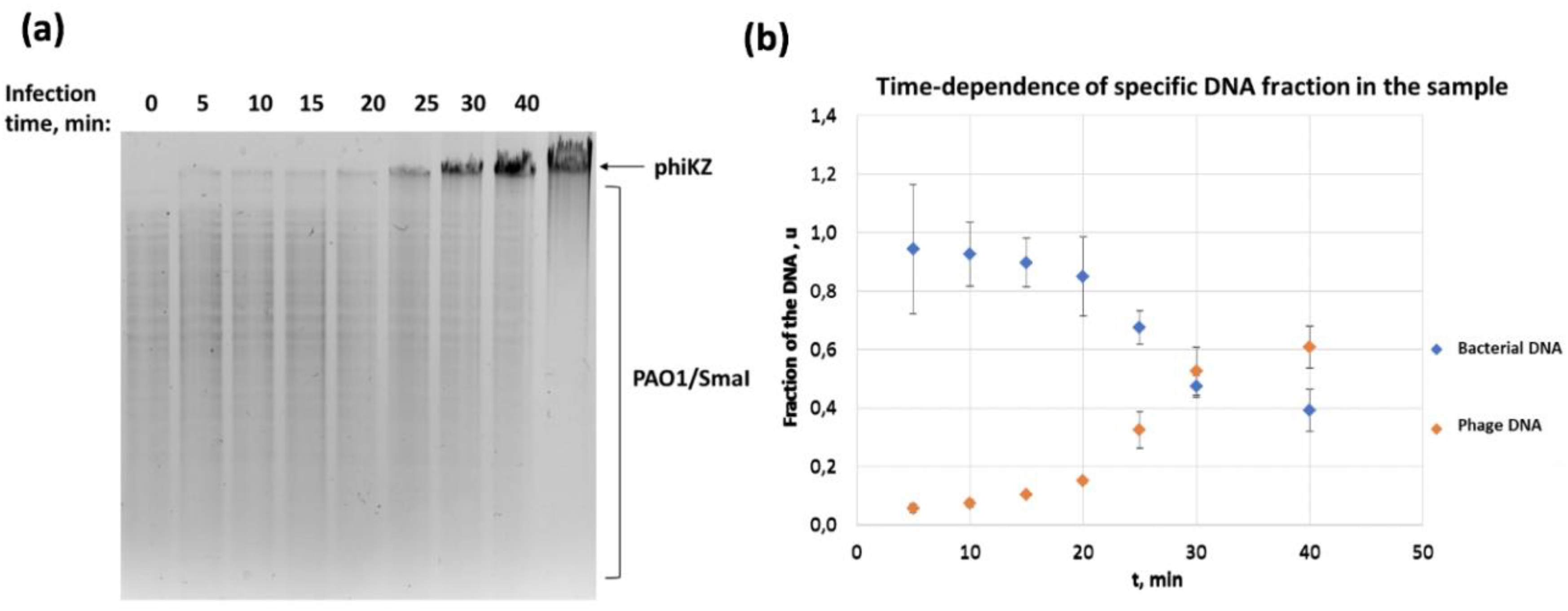
3.2. Concentrations of Phage and Bacterial DNA Switch in the Course of phiKZ Infection
3.3. Localization of Bacterial and Phage DNA During Infection
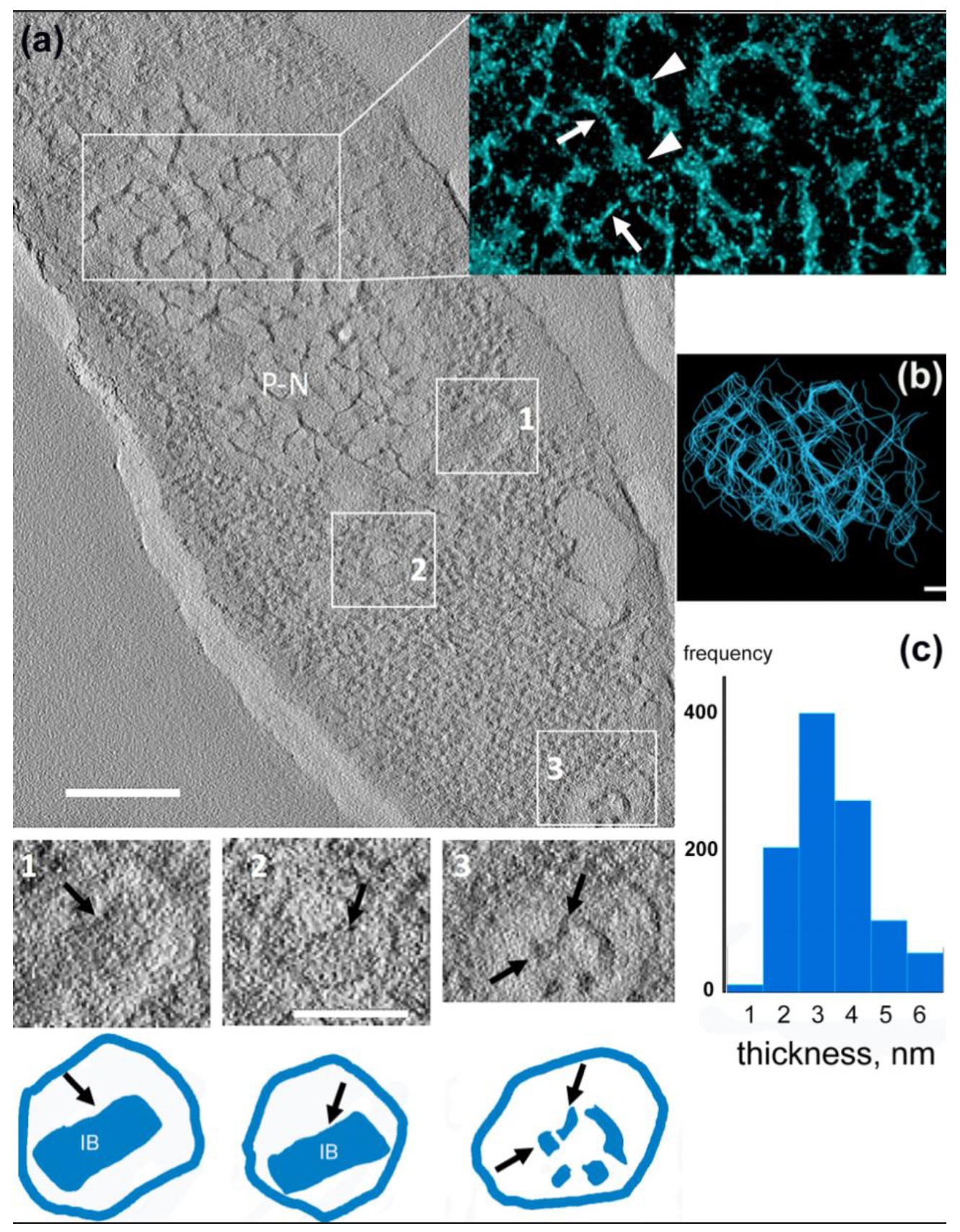
3.4. 3D Structure of the Pseudo-Nucleus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hertveldt, K.; Lavigne, R.; Pleteneva, E.; Sernova, N.; Kurochkina, L.; Korchevskii, R.; Robben, J.; Mesyanzhinov, V.; Krylov, V.N.; Volckaert, G. Genome comparison of pseudomonas aeruginosa large phages. J. Mol. Biol. 2005, 354, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Mesyanzhinov, V.V.; Robben, J.; Grymonprez, B.; Kostyuchenko, V.A.; Bourkaltseva, M.V.; Sykilinda, N.N.; Krylov, V.N.; Volckaert, G. The genome of bacteriophage φKZ of Pseudomonas aeruginosa. J. Mol. Biol. 2002, 317, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ceyssens, P.-J.; Minakhin, L.; Van den Bossche, A.; Yakunina, M.; Klimuk, E.; Blasdel, B.; De Smet, J.; Noben, J.-P.; Blasi, U.; Severinov, K.; et al. Development of giant bacteriophage ϕKZ is independent of the host transcription apparatus. J. Virol. 2014, 88, 10501–10510. [Google Scholar] [CrossRef] [PubMed]
- Yakunina, M.; Artamonova, T.; Borukhov, S.; Makarova, K.S.; Severinov, K.; Minakhin, L. A non-canonical multisubunit RNA polymerase encoded by a giant bacteriophage. Nucleic Acids Res. 2015, gkv1095. [Google Scholar] [CrossRef] [PubMed]
- Lecoutere, E.; Ceyssens, P.-J.; Miroshnikov, K.A.; Mesyanzhinov, V.V.; Krylov, V.N.; Noben, J.-P.; Robben, J.; Hertveldt, K.; Volckaert, G.; Lavigne, R. Identification and comparative analysis of the structural proteomes of ϕKZ and EL, two giant Pseudomonas aeruginosa bacteriophages. Proteomics 2009, 9, 3215–3219. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Rolando, M.R.; Carroll, C.A.; Shen, P.S.; Belnap, D.M.; Weintraub, S.T.; Serwer, P.; Hardies, S.C. Characterization of Pseudomonas chlororaphis myovirus 201ϕ2-1 via genomic sequencing, mass spectrometry, and electron microscopy. Virology 2008, 376, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Krylov, V.N.; Smirnova, T.A.; Minenkova, I.B.; Plotnikova, T.G.; Zhazikov, I.Z.; Khrenova, E.A. Pseudomonas bacteriophage contains an inner body in its capsid. Can. J. Microbiol. 1984, 30, 758–762. [Google Scholar] [CrossRef]
- Sokolova, O.S.; Shaburova, O.V.; Pechnikova, E.V.; Shaytan, A.K.; Krylov, S.V.; Kiselev, N.A.; Krylov, V.N. Genome packaging in EL and Lin68, two giant phiKZ-like bacteriophages of P. aeruginosa. Virology 2014, 468–470. [Google Scholar] [CrossRef]
- Matsko, N.; Klinov, D.; Manykin, A.; Demin, V.; Klimenko, S. Atomic force microscopy analysis of bacteriophages phiKZ and T4. J. Electron Microsc. 2001, 50, 417–422. [Google Scholar] [CrossRef]
- Burrowes, B.; Harper, D.R.; Anderson, J.; McConville, M.; Enright, M.C. Bacteriophage therapy: Potential uses in the control of antibiotic-resistant pathogens. Expert Rev. Anti-Infect. Ther. 2011, 9, 775–785. [Google Scholar] [CrossRef]
- Chang, R.Y.K.; Wallin, M.; Lin, Y.; Leung, S.S.Y.; Wang, H.; Morales, S.; Chan, H.-K. Phage therapy for respiratory infections. Adv. Drug Deliv. Rev. 2018, 133, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Torres-Barceló, C.; Arias-Sánchez, F.I.; Vasse, M.; Ramsayer, J.; Kaltz, O.; Hochberg, M.E. A window of opportunity to control the bacterial pathogen pseudomonas aeruginosa combining antibiotics and phages. PLoS ONE 2014, 9, e106628. [Google Scholar] [CrossRef] [PubMed]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Meselson, M.; Yuan, R.; Heywood, J. Restriction and modification of DNA. Annu. Rev. Biochem. 1972, 41, 447–466. [Google Scholar] [CrossRef]
- Mendoza, S.D.; Nieweglowska, E.S.; Govindarajan, S.; Leon, L.M.; Berry, J.D.; Tiwari, A.; Chaikeeratisak, V.; Pogliano, J.; Agard, D.A.; Bondy-Denomy, J. A bacteriophage nucleus-like compartment shields DNA from CRISPR nucleases. Nature 2020, 577, 244–248. [Google Scholar] [CrossRef]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef]
- Fernández, L.; Rodríguez, A.; García, P. Phage or foe: An insight into the impact of viral predation on microbial communities. ISME J. 2018, 12, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Campoy, S.; Hervàs, A.; Busquets, N.; Erill, I.; Teixidó, L.; Barbé, J. Induction of the SOS response by bacteriophage lytic development in Salmonella enterica. Virology 2006, 351, 360–367. [Google Scholar] [CrossRef][Green Version]
- Fallico, V.; Ross, R.P.; Fitzgerald, G.F.; McAuliffe, O. Genetic response to bacteriophage infection in lactococcus lactis reveals a four-strand approach involving induction of membrane stress proteins, D-alanylation of the cell wall, maintenance of proton motive force, and energy conservation. J. Virol. 2011, 85, 12032–12042. [Google Scholar] [CrossRef]
- Ainsworth, S.; Zomer, A.; Mahony, J.; van Sinderen, D. Lytic infection of lactococcus lactis by bacteriophages Tuc2009 and c2 triggers alternative transcriptional host responses. Appl. Environ. Microbiol. 2013, 79, 4786–4798. [Google Scholar] [CrossRef]
- Gao, B.L.; Altae-Tran, H.; Böhning, F.; Makarova, K.S.; Segel, M.; Schmid-Burgk, J.L.; Koob, J.; Wolf, Y.L.; Koonin, E.V.; Zhang, F. Diverse enzymatic activities mediate antiviral immunity in prokaryotes. Science 2020, 369, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Cornuault, J.K.; Moineau, S. A jumbo formation in the viral game plan. CRISPR J. 2020, 3, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Chaikeeratisak, V.; Nguyen, K.; Egan, M.E.; Erb, M.L.; Vavilina, A.; Pogliano, J. The phage nucleus and tubulin spindle are conserved among large pseudomonas phages. Cell Rep. 2017, 20, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Chaikeeratisak, V.; Nguyen, K.; Khanna, K.; Brilot, A.F.; Erb, M.L.; Coker, J.K.C.; Vavilina, A.; Newton, G.L.; Buschauer, R.; Pogliano, K.; et al. Assembly of a nucleus-like structure during viral replication in bacteria. Science 2017, 355, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Mastronarde, D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005, 152, 36–51. [Google Scholar] [CrossRef]
- Kremer, J.R.; Mastronarde, D.N.; McIntosh, J.R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996, 116, 71–76. [Google Scholar] [CrossRef]
- Introduction to 3dmod, Version 4.9. Available online: https://bio3d.colorado.edu/imod/doc/3dmodguide.html (accessed on 28 July 2019).
- Loiko, N.; Danilova, Y.; Moiseenko, A.; Kovalenko, V.; Tereshkina, K.; Tutukina, M.; El-Registan, G.; Sokolova, O.; Krupyanskii, Y. Morphological peculiarities of the DNA-protein complexes in starved Escherichia coli cells. PLoS ONE 2020, 15, e0231562. [Google Scholar] [CrossRef]
- Fokine, A.; Battisti, A.J.; Bowman, V.D.; Efimov, A.V.; Kurochkina, L.P.; Chipman, P.R.; Mesyanzhinov, V.V.; Rossmann, M.G. Cryo-EM study of the pseudomonas bacteriophage φKZ. Structure 2007, 15, 1099–1104. [Google Scholar] [CrossRef]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Lewin, B. Genes IX, 9th ed.; Jones & Bartlett Publishers: Sudbury, MA, USA, 2007. [Google Scholar]
- Wu, W.; Thomas, J.A.; Cheng, N.; Black, L.W.; Steven, A.C. Bubblegrams reveal the inner body of bacteriophage ϕKZ. Science 2012, 335, 182. [Google Scholar] [CrossRef]
- Erb, M.L.; Kraemer, J.A.; Coker, J.K.C.; Chaikeeratisak, V.; Nonejuie, P.; Agard, D.A.; Pogliano, J. A bacteriophage tubulin harnesses dynamic instability to center DNA in infected cells. Elife 2014, 3. [Google Scholar] [CrossRef]
- Reilly, E.R.; Abajorga, M.K.; Kiser, C.; Mohd Redzuan, N.H.; Haidar, Z.; Adams, L.E.; Diaz, R.; Pinzon, J.A.; Hudson, A.O.; Black, L.W.; et al. A Cut above the Rest: Characterization of the Assembly of a Large Viral Icosahedral Capsid. Viruses 2020, 12, 725. [Google Scholar] [CrossRef] [PubMed]
- Trinh, J.T.; Shao, Q.; Guan, J.; Zeng, L. Emerging heterogeneous compartments by viruses in single bacterial cells. Nat. Commun. 2020, 11, 3813. [Google Scholar] [CrossRef] [PubMed]
- Frenkiel-Krispin, D.; Minsky, A. Nucleoid organization and the maintenance of DNA integrity in E. coli, B. subtilis and D. radiodurans. J. Struct. Biol. 2006, 156, 311–319. [Google Scholar] [CrossRef]
- Almirón, M.; Link, A.J.; Furlong, D.; Kolter, R. A novel DNA-binding protein with regulatory and protective roles in starved Escherichia coli. Genes Dev. 1992, 6, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Minsky, A.; Shimoni, E.; Frenkiel-Krispin, D. Stress, order and survival. Nat. Rev. Mol. Cell Biol. 2002, 3, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Hanninen, A.-L.; Bamford, D.H.; Bamford, J.K.H. Assembly of membrane-containing bacteriophage PRD1 is dependent on GroEL and GroES. Virology 1997, 227, 207–210. [Google Scholar] [CrossRef]
- De Smet, J.; Zimmermann, M.; Kogadeeva, M.; Ceyssens, P.-J.; Vermaelen, W.; Blasdel, B.; Bin Jang, H.; Sauer, U.; Lavigne, R. High coverage metabolomics analysis reveals phage-specific alterations to Pseudomonas aeruginosa physiology during infection. ISME J. 2016, 10, 1823–1835. [Google Scholar] [CrossRef]
- Moiseenko, A.; Loiko, N.; Tereshkina, K.; Danilova, Y.; Kovalenko, V.; Chertkov, O.; Feofanov, A.V.; Krupyanskii, Y.F.; Sokolova, O.S. Projection structures reveal the position of the DNA within DNA-Dps Co-crystals. Biochem. Biophys. Res. Commun. 2019, 517, 463–469. [Google Scholar] [CrossRef]
- Frenkiel-Krispin, D.; Ben-Avraham, I.; Englander, J.; Shimoni, E.; Wolf, S.G.; Minsky, A. Nucleoid restructuring in stationary-state bacteria. Mol. Microbiol. 2004, 51, 395–405. [Google Scholar] [CrossRef]
- Wolf, S.G.; Frenkiel, D.; Arad, T.; Finkel, S.E.; Kolter, R.; Minsky, A. DNA protection by stress-induced biocrystallization. Nature 1999, 400, 83–85. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fraction of Infected Cells in Culture, x | Estimated Value of the DNAs Mass Ratio | Experimental Value of the DNAs Mass Ratio |
|---|---|---|
| 0.8 | 0.036 | 0.062 |
| 0.65 | 0.029 | 0.073 |
| Fraction of Infected Cells in Culture, x | Estimated Value of DNAs Mass Ratio without Bacterial DNA Degradation | Estimated Value of DNAs Mass Ratio with Full Bacterial DNA Degradation | Experimental Value of DNAs Mass Ratio |
|---|---|---|---|
| 0.8 | 2.03 | 10.17 | 1.55 |
| 0.65 | 1.65 | 4.72 | 0.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danilova, Y.A.; Belousova, V.V.; Moiseenko, A.V.; Vishnyakov, I.E.; Yakunina, M.V.; Sokolova, O.S. Maturation of Pseudo-Nucleus Compartment in P. aeruginosa, Infected with Giant phiKZ Phage. Viruses 2020, 12, 1197. https://doi.org/10.3390/v12101197
Danilova YA, Belousova VV, Moiseenko AV, Vishnyakov IE, Yakunina MV, Sokolova OS. Maturation of Pseudo-Nucleus Compartment in P. aeruginosa, Infected with Giant phiKZ Phage. Viruses. 2020; 12(10):1197. https://doi.org/10.3390/v12101197
Chicago/Turabian StyleDanilova, Yana A., Viktoriia V. Belousova, Andrey V. Moiseenko, Innokentii E. Vishnyakov, Maria V. Yakunina, and Olga S. Sokolova. 2020. "Maturation of Pseudo-Nucleus Compartment in P. aeruginosa, Infected with Giant phiKZ Phage" Viruses 12, no. 10: 1197. https://doi.org/10.3390/v12101197
APA StyleDanilova, Y. A., Belousova, V. V., Moiseenko, A. V., Vishnyakov, I. E., Yakunina, M. V., & Sokolova, O. S. (2020). Maturation of Pseudo-Nucleus Compartment in P. aeruginosa, Infected with Giant phiKZ Phage. Viruses, 12(10), 1197. https://doi.org/10.3390/v12101197