1. Introduction
The polyomaviruses are small, non-enveloped, double-stranded DNA tumor viruses that include pathogenic human viruses such as BK polyomavirus (BKPyV), JC polyomavirus (JCPyV), and Merkel Cell polyomavirus (MCPyV) as well as the extensively studied model viruses, murine polyomavirus and simian virus 40 (SV40). MCPyV, the most recently discovered human tumor virus, is responsible for most cases of Merkel cell carcinoma, a rare but aggressive form of skin cancer. BKPyV is associated with inflammation of the urogenital tract and nephropathy, which can result in organ loss in renal transplant patients as well as hemorrhagic cystitis in bone marrow transplant recipients (reviewed in [
1]). JCPyV is the causative agent of progressive multifocal leukencephalopathy (PML), a rare but usually fatal central nervous system-demyelinating disease in immunocompromised individuals or patients receiving immunomodulatory monoclonal antibody treatment for various disorders [
2]. JCPyV and BKPyV infections are common in the human population. These viruses are phylogenetically closely related to SV40, which can cause PML-like brain pathology in immunosuppressed monkeys [
3,
4]. Therefore, SV40 serves as a model to study human polyomavirus pathogenesis, including neurological disease.
Productive polyomavirus infection of permissive cells can be divided into early and late phases. The early phase includes virus binding to the cell surface, entry of virus particles into the cell, and trafficking of the viral genome to the nucleus where viral gene expression and DNA replication occur. Polyomavirus entry is initiated by binding of the major capsid protein VP1 to carbohydrate motifs on cell surface molecules. In the case of SV40, the ganglioside GM1 serves as the cellular receptor for infection. After endocytosis, polyomaviruses are transported to the endoplasmic reticulum (ER), where host factors initiate the disassembly of capsids and translocation of the viral genome and residual capsid into the cytoplasm for transport into the nucleus [
5,
6]. Subsequent expression of the early viral proteins including large and small T antigens is followed by viral DNA replication, expression of the late viral proteins including VP1, and capsid assembly, which occurs primarily in the nucleus before cells are lysed and mature infectious virus particles are released.
The initial interaction of a variety of polyomaviruses with cells acutely induces transient cellular signaling that supports the early steps of infection. The JC virus induces ERK phosphorylation within minutes after receptor binding [
7], which is required for the early stages of infection [
8]. Within the first two hours of infection, murine polyomavirus induces the phosphoinositide 3′ kinase and Fak signaling pathways through binding of VP1 to gangliosides and alpha4-integrin [
9,
10,
11]. Inhibition of these signaling events can inhibit the early steps of murine polyomavirus infection. This virus also induces a second, delayed wave of mitogenic signaling that depends on viral early gene expression [
11]. Cell signaling also modulates productive SV40 infection [
12]. Binding of SV40 to GM1 at the plasma membrane triggers activation of more than 50 different kinases regulating the early steps of SV40 infection including local activation of tyrosine kinases to reorganize actin filaments for caveolin-1- or lipid raft-dependent SV40 internalization [
13].
In contrast to the early stages of polyomavirus infection, late events leading to the release of virus particles are poorly understood. Viral proteins have been reported to facilitate SV40 release from cells. The late protein VP4 has been reported to function as a viroporin with membrane-destabilizing properties that facilitates virus release, but these results have recently been challenged [
14,
15,
16,
17]. Furthermore, since VP4 is mostly found within the nucleus of infected cells, the mechanism leading to plasma membrane perforation and virus release is unclear. The minor capsid proteins VP2 and VP3 were also reported to support membrane permeabilization for virus release [
14]. These proteins can insert into or disrupt membranes when ectopically overexpressed in prokaryotic as well as eukaryotic cells.
SV40 infection of African green monkey cells leads to the appearance of characteristic cytoplasmic vacuoles late during infection, a phenomenon that led to the discovery of this virus in 1960 [
18]. We showed that vacuolization is triggered by the interaction between VP1 and GM1 at the cell surface [
19,
20]. SV40-induced vacuolization typically occurs late in infection, but if large amounts of SV40 are added to cells, vacuoles can form acutely [
19,
21]. Virus replication is not required for vacuole formation, and purified VP1 pentamers are sufficient to induce vacuole formation. We hypothesized that the VP1–GM1 interaction triggers an as-yet-unidentified signaling cascade resulting in vacuolization [
19].
Extensive cell vacuolization has also been observed in other experimental systems. Pore-forming toxins of various pathogens can induce the formation of cellular vacuoles and cell death [
22,
23]. Different types of intrinsic cell death programs, such as paraptosis and methuosis, are also associated with vacuole formation [
22,
24]. Cell signaling pathways including the Ras-MAPK pathway have been shown to contribute to vacuolization and nonapoptotic cell lysis in these processes [
24,
25]. However, cellular factors or signaling pathways have not been identified that are involved in vacuolization or other late events during SV40 infection.
In this study, we investigate the mechanism by which SV40 infection results in efficient virus release. We show that activation of the Ras-Rac1-MKK4-JNK signaling pathway late during SV40 infection results in vacuolization and ultimately facilitates cell lysis and release of progeny virus. Understanding the mechanism of polyomavirus release may allow the identification of proteins and pathways that can potentially be exploited as specific antiviral drug targets for polyomaviruses and possibly other pathogenic, non-enveloped viruses.
2. Materials and Methods
2.1. Cells and Virus
CV-1 cells and SV40 776 virus DNA were purchased from American Type Culture Collection (ATCC). Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS), 10 mM L-glutamine, and 10 mM HEPES (pH 7.2) in 5% CO2 at 37 °C. SV40 was produced from SV40 776 in a bacterial vector backbone puc19 by excision, religation, and transfection into CV-1 cells. When significant cell death was observed, cell cultures were subjected to multiple rounds of freeze/thaw lysis. Cellular debris was removed by centrifugation at 1000 rpm for 5 min, and supernatants were filtered through 0.45-µm syringe filters, aliquoted, and stored at −80 °C. To produce higher titer virus stocks, fresh CV-1 cells were infected at an MOI of 0.5 and processed as described above.
2.2. Virus Titer Quantitation
To quantify infectious units of SV40, serial dilutions of virus preparation, tissue culture supernatant, or cell lysate were added to monolayers of 2 × 105 CV-1 cells in six-well plates. After 24 h, CV-1 cells were trypsinized, fixed, and permeabilized in methanol or with 4% PFA/0.5% Triton X-100 before being subjected to immunofluorescence staining for large T antigen and flow cytometry. In a typical infection with wildtype SV40, at 48 h.p.i., there is approximately 5- to 9-fold more virus in the supernatant than in the cell lysate.
2.3. Immunoblots
CV-1 cells were seeded at 2 × 105 in six-well plates and infected with SV40 on the following day at an MOI of 10. At indicated times p.i., cells were harvested by lysis in lysis buffer (2% Triton X-100, 0.5% Na-deoxycholate, 150 mM NaCl, 25 mM Tris, 5 mM EDTA, halt protease, and phosphatase inhibitors (Thermo Scientific, Waltham, MA, USA )). Extracts were suspended in 5 × Laemmli buffer, subjected to sonication, and boiled. Equal sample volumes were loaded on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) for protein separation. Proteins were then transferred to 0.2-μm polyvinylidene difluoride (PVDF) membranes in Tris/glycine transfer buffer (25 mM Tris, 192 mM glycine, and 20% methanol) for 2 h at 100 V. Membranes were blocked in 5% bovine serum albumin (BSA) or non-fat dry milk in Tris-buffered saline with Tween-20 (TBST) (10 mM Tris-HCl, pH 7.4, 167 mM NaCl, and 1% Tween-20) for 1 h and incubated overnight at 4 °C with indicated antibodies in 5% BSA/TBST for phosphorylated targets or in 5% non-fat dry milk/TBST for all others. Blots were washed in TBST and incubated for 1 h at room temperature with HRP-conjugated donkey anti-mouse/rabbit/goat (Jackson ImmunoResearch, Philadelphia, PA, USA) in 5% non-fat dry milk/TBST. After washing with TBST, blots were visualized by enhanced chemiluminescence (SuperSignal West Pico/Femto Chemiluminescent Substrate, Thermo Scientific). The following primary antibodies were used for immunoblotting: anti-ß-actin (Abcam, #ab8227), anti-p-ERK (Cell Signaling Technology, CST, #4370), anti-p-JNK (CST, #4668), anti-p-p38 (CST, #4511), anti-p-MKK4 (CST, #9156), anti-ERK (CST, #4695), anti-JNK (CST, #9252), anti-p38 (CST, #8690), anti-p-ATF2 (CST, #9225), anti-p-cJun (CST, #3270), anti-large T antigen (PAb, #108), and anti-VP1 (PAb, #597).
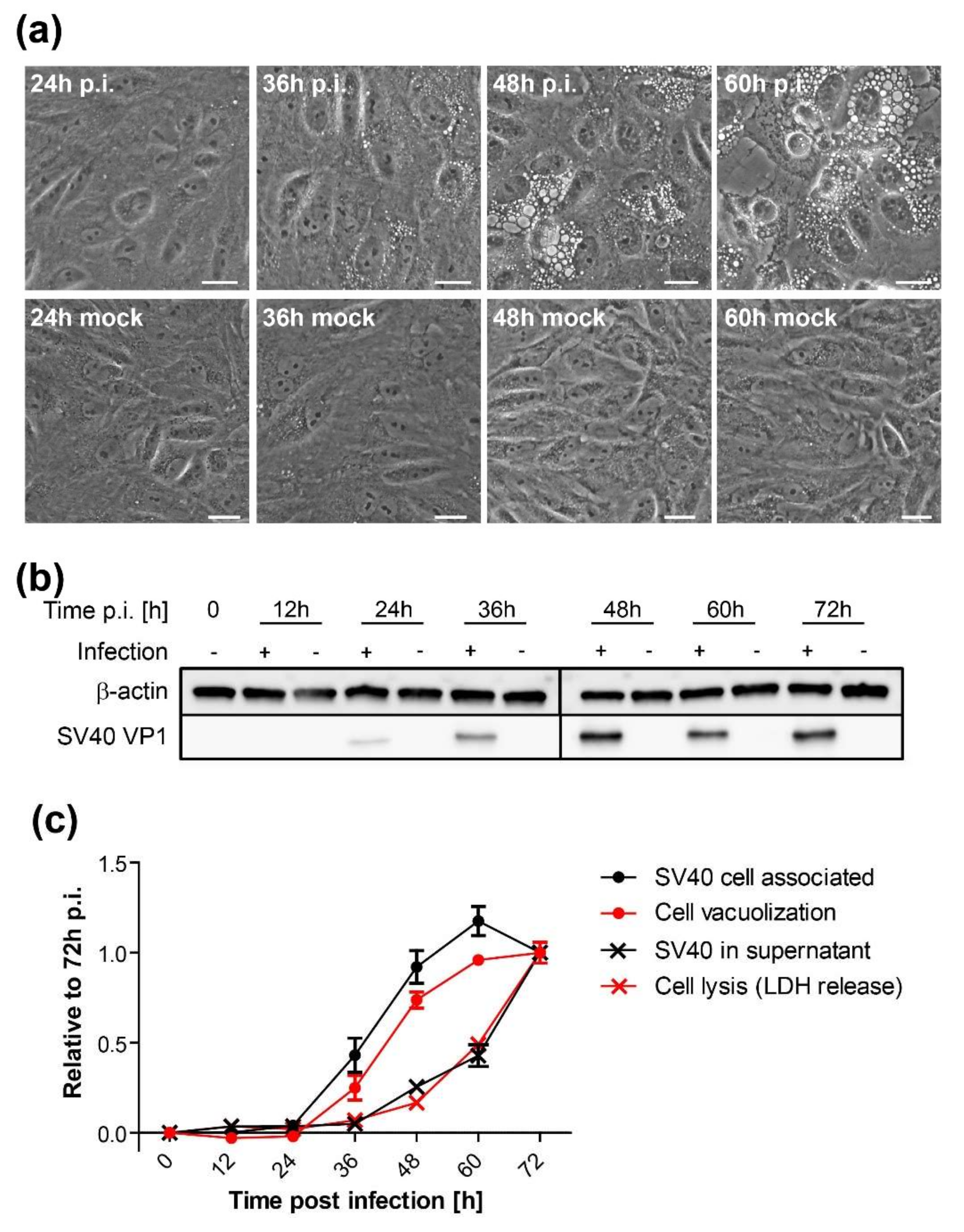
2.4. Vacuolization Assay
CV-1 cells were plated at a density of 2 × 105 cells per well on six-well plates and infected with SV40 on the following day at an MOI of 10. At indicated time points, vacuolization was assessed and documented by bright-field microscopy. Only cells containing vacuoles with a diameter larger than 5 µm were counted positive. Mock-treated cells were initially used as a reference to differentiate membrane vesicles of the endosomal system from SV40-induced vacuoles. A minimum of 200 cells per sample were counted under blinded conditions.
2.5. Chemical Inhibitor Treatment
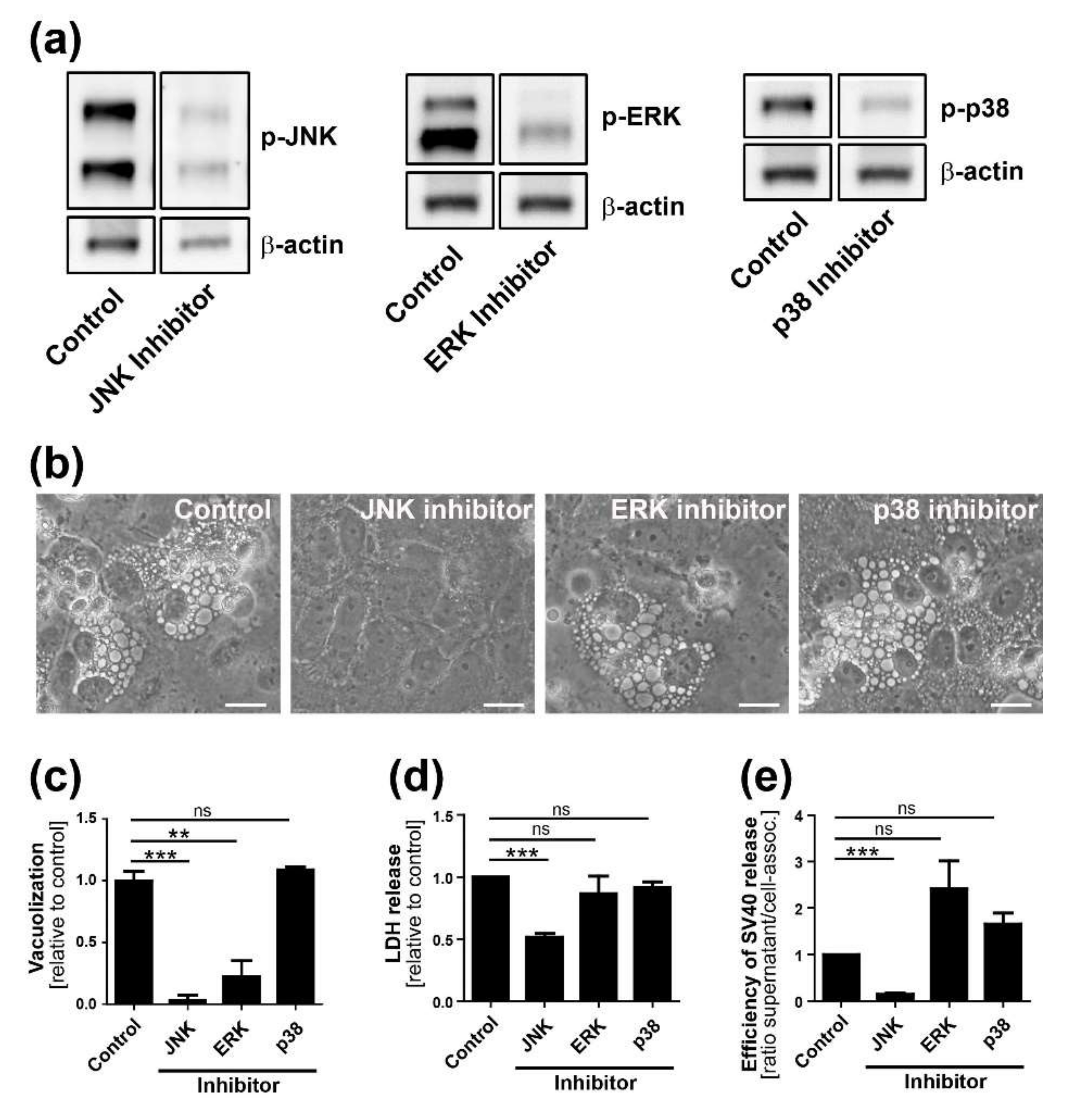
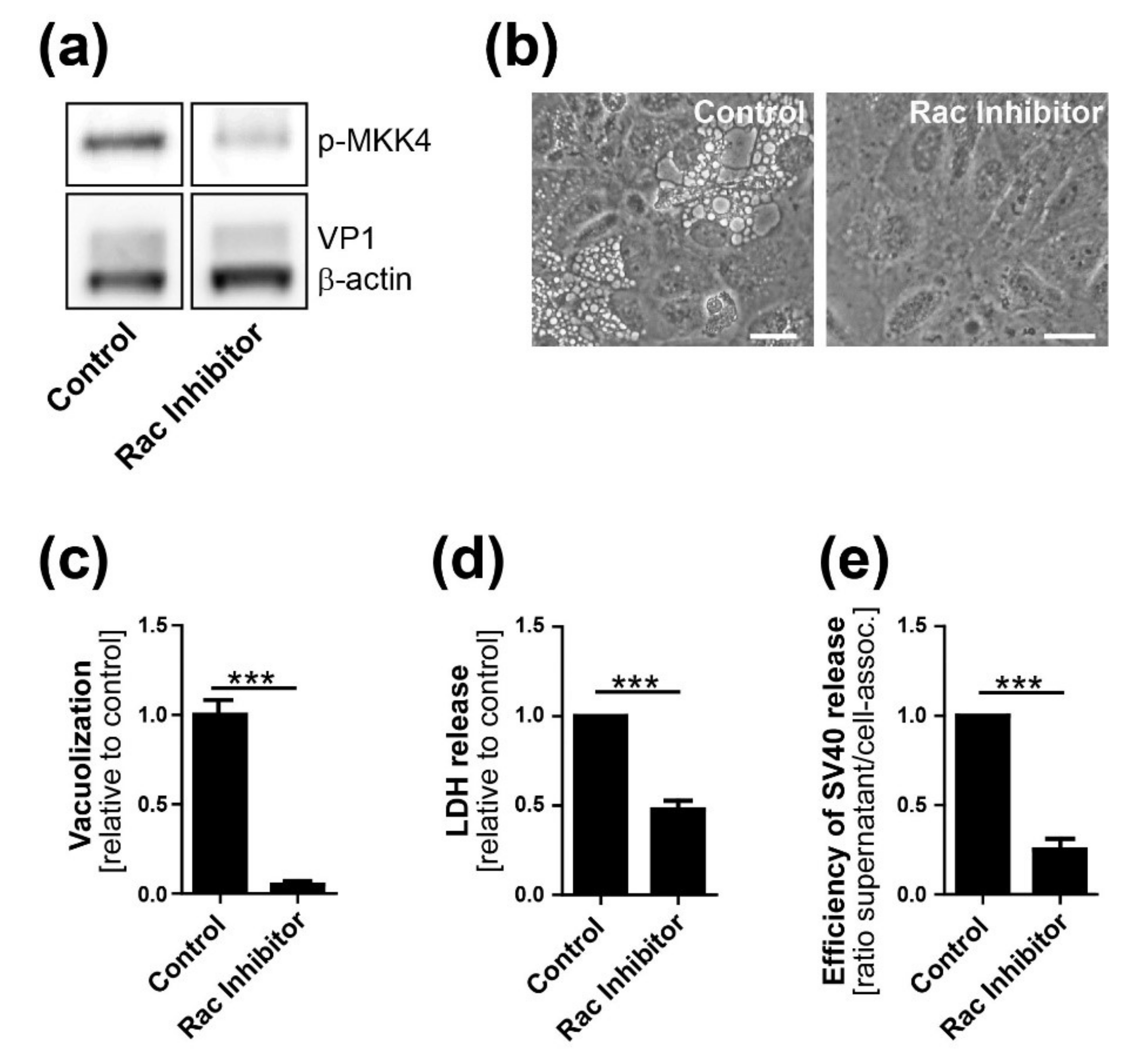
CV-1 cells were plated at a density of 2 × 105 on six-well plates. On the following day, cells were infected with SV40 at an MOI of 10. Twelve h.p.i. or at the time of infection (0 h.p.i.), chemical inhibitors were added at a concentration of 25 µM to the infected and mock-treated CV-1 cells. Inhibitor treatment was maintained until the end of the experiment. The following chemical inhibitors were used in this study: Selumetinib (ERK pathway inhibitor, inhibits MEK1), EHT 1864 (Rac inhibitor), SB203580 (p38 inhibitor), and SP600125 (JNK inhibitor). All inhibitors were purchased from Selleckchem (Houston, TX, USA).
2.6. qRT-PCR
For quantitative PCR, total RNA was extracted from 2 × 105 cells using the RNEasy Mini Kit (Qiagen, Hilden, Germany) and a maximum of 1 µg cDNA was transcribed with the iScript™ cDNA Synthesis Kit (BioRad, CA, USA). The relative expression levels were assessed in triplicate on a single-color detection system (BioRad CFX Connect Real-Time PCR Detection System) with the iTaq™ universal SYBR Green supermix (BioRad, CA, USA). Genes and primers used for qPCR were as follows: GAPDH forward (TGGTATCGTGGAAGGACTCA), GAPDH reverse (CCAGTAGAGGCAGGGATGAT), MKK4 forward (TGAAAAGGCACAAAGTAAACGCA), and MKK4 reverse (CCCAGTGTTGTTCAGGGGAG).
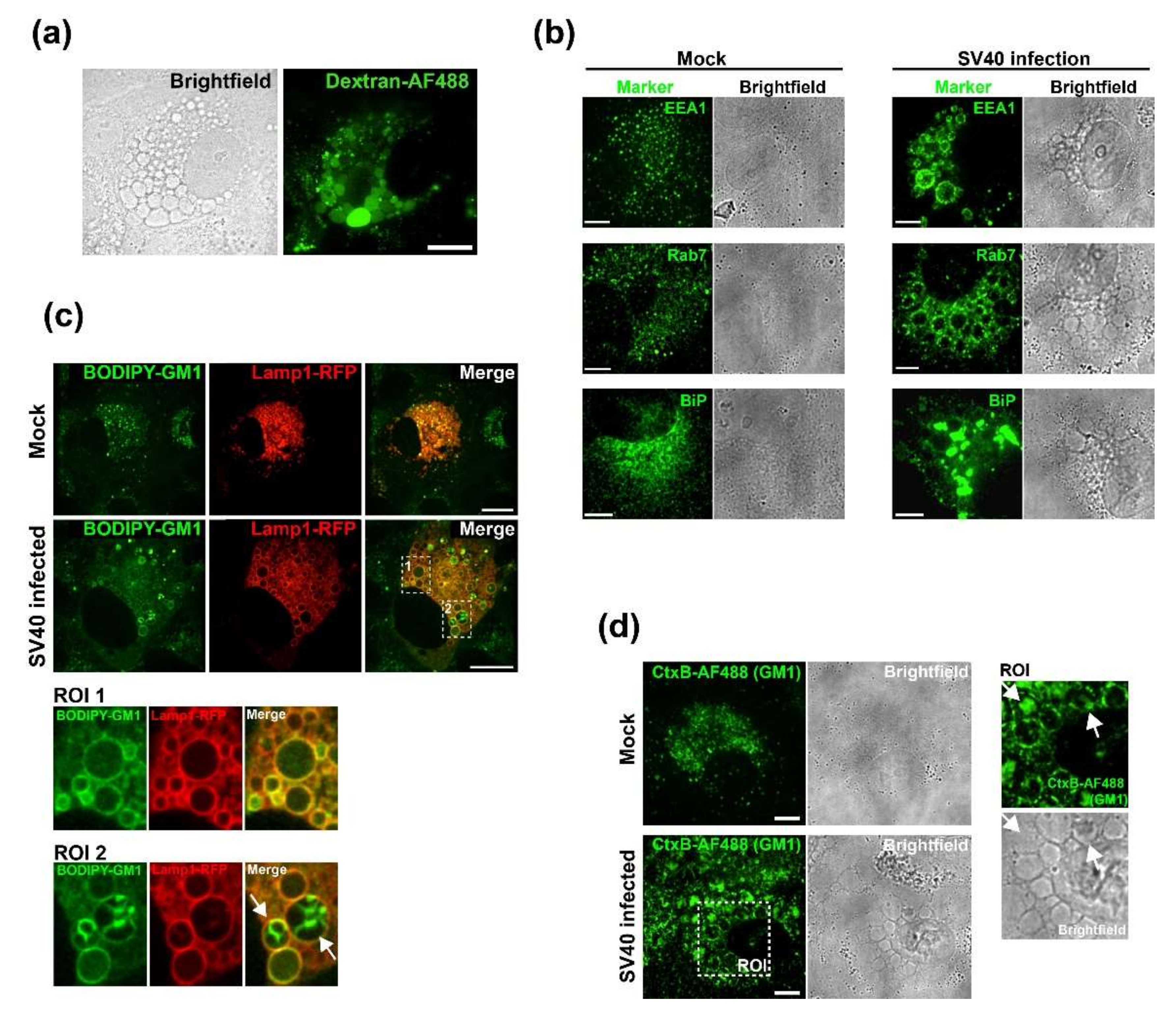
2.7. Dextran and BODIPY-GM1 Treatment
CV-1 cells were plated at a density of 2 × 104 on Lab-Tek II chambered coverglass slides (Nunc) and infected with SV40 at an MOI of 100 on the following day. At 47 h.p.i., cells were incubated for 1 h with fluorescent markers. For dextran uptake assays, the cell culture medium was supplemented with 0.25 mg/mL of 3 kDa Dextran conjugated with Alexa Fluor 488 (Dextran-AF488, Life Technologies, USA). For labeling with fluorescent GM1, 5 µM BODIPY FL C5-GM1 (Molecular Probes) was added to the cell culture medium. At 48 h.p.i., cells were thoroughly washed and fresh cell culture medium was added for imaging. Cells were imaged at 50 h.p.i. on a Nikon TE2000 spinning disk confocal microscope driven by the Volocity software package (Perkin Elmer, Hopkinton, MA, USA).
2.8. Immunofluorescence
CV-1 cells were plated at a density of 3 × 104 on Millicell EZ Slide four-well glass slides (Millipore, USA). On the following day, cells were infected with SV40 at an MOI of 100. After 48 h, cells were fixed with 4% PFA, permeabilized with 0.5% Triton X-100, and immunostained or treated with 0.5 µg/mL Alexa Fluor 488 (AF488)-conjugated CtxB (Molecular Probes) to stain GM1. The following primary antibodies were used for immunofluorescence staining: anti-VP1 (Abcam, #ab53977), anti-EEA1 (CST, #C45B10), anti-Rab7 (CST, #D95F2), and anti-BiP (Abcam, #ab108615). The secondary antibodies donkey anti-mouse/anti-rabbit conjugated to Alexa Fluor-488 or -568 (Life Technologies, USA) were used. Stained samples were embedded in ProLong Gold (Invitrogen, Waltham, MA, USA), and data were acquired on a spinning disk confocal microscope (Nikon, Japan). Images were analyzed using Volocity software (PerkinElmer).
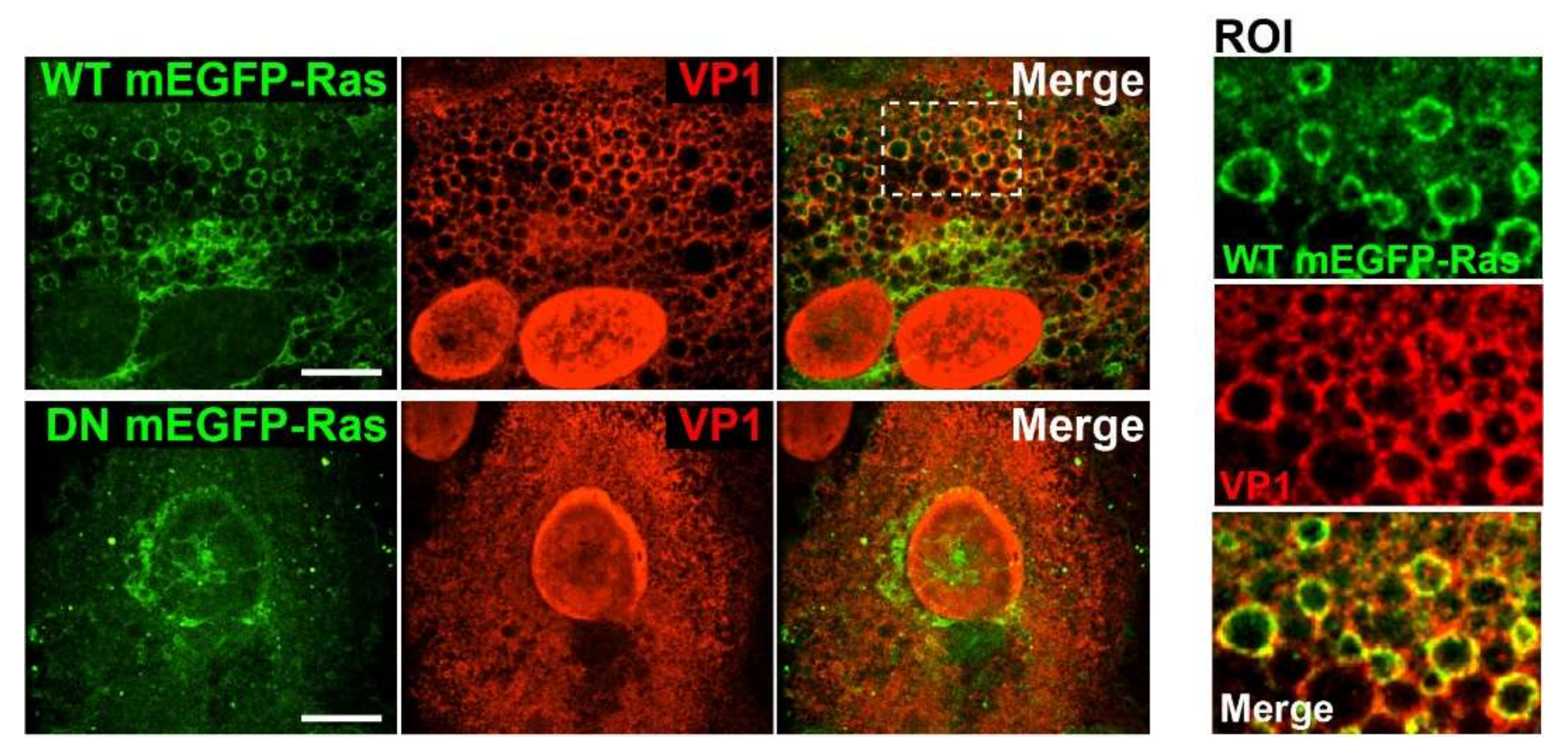
For experiments involving Ras expression, 2 × 105 CV-1 cells were plated on coverslips in six-well plates. Cells were transfected with wildtype (WT) or dominant-negative (DN) mEGFP-HRas using FuGENE6 (Promega, Madison, WI, USA) transfection reagent. On the following day, cells were infected with SV40 at an MOI of 10. At 48 h.p.i., the cells were fixed and immunostained with a primary antibody against anti-VP1 (Abcam, Cambridge, UK) and the secondary antibody anti-rabbit Alexa Fluor 568. Stained samples were embedded in ProLong Gold (Invitrogen, MA, USA), and data were acquired on a spinning disk confocal microscope (Nikon, Japan). Images were analyzed using Volocity software (PerkinElmer, MA, USA). The plasmids encoding WT mEGFP-HRas (Plasmid #18662) and DN mEGFP-HRas S17N (Plasmid #18665) were purchased from Addgene (Watertown, Massachusetts, USA).
2.9. Live-Cell Microscopy
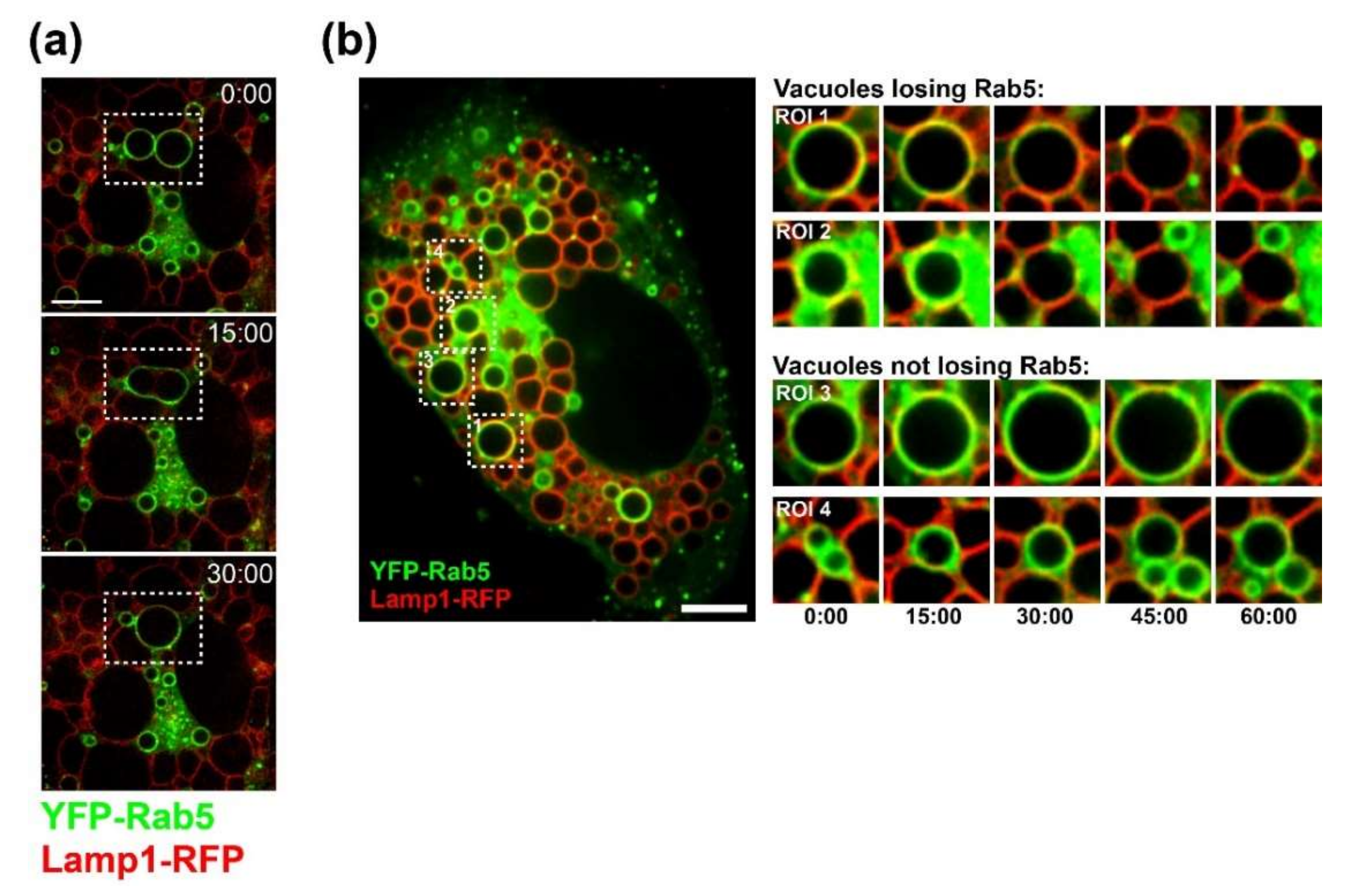
CV-1 cells were plated at a density of 1.5 × 104 on Lab-Tek II chambered coverglass slides (Nunc). On the following day, cells were infected with SV40 at an MOI of 100. At 20 h.p.i., cells were co-transfected with plasmids encoding Lamp1-RFP and YFP-Rab5 using FuGENE6 (Promega, WI, USA) transfection reagent. Time-lapse microscopy started at 40 h.p.i. Images were acquired every 15 min on a Nikon spinning disk confocal microscope with Nikon perfect focus system and a LiveCell environmental chamber (Pathology Devices, Westminster, CA, USA). Volocity software (PerkinElmer, MA, USA) and ImageJ were used for 4D image analysis.
2.10. Generation of MKK4 Knockdown CV-1 Cells with shRNA
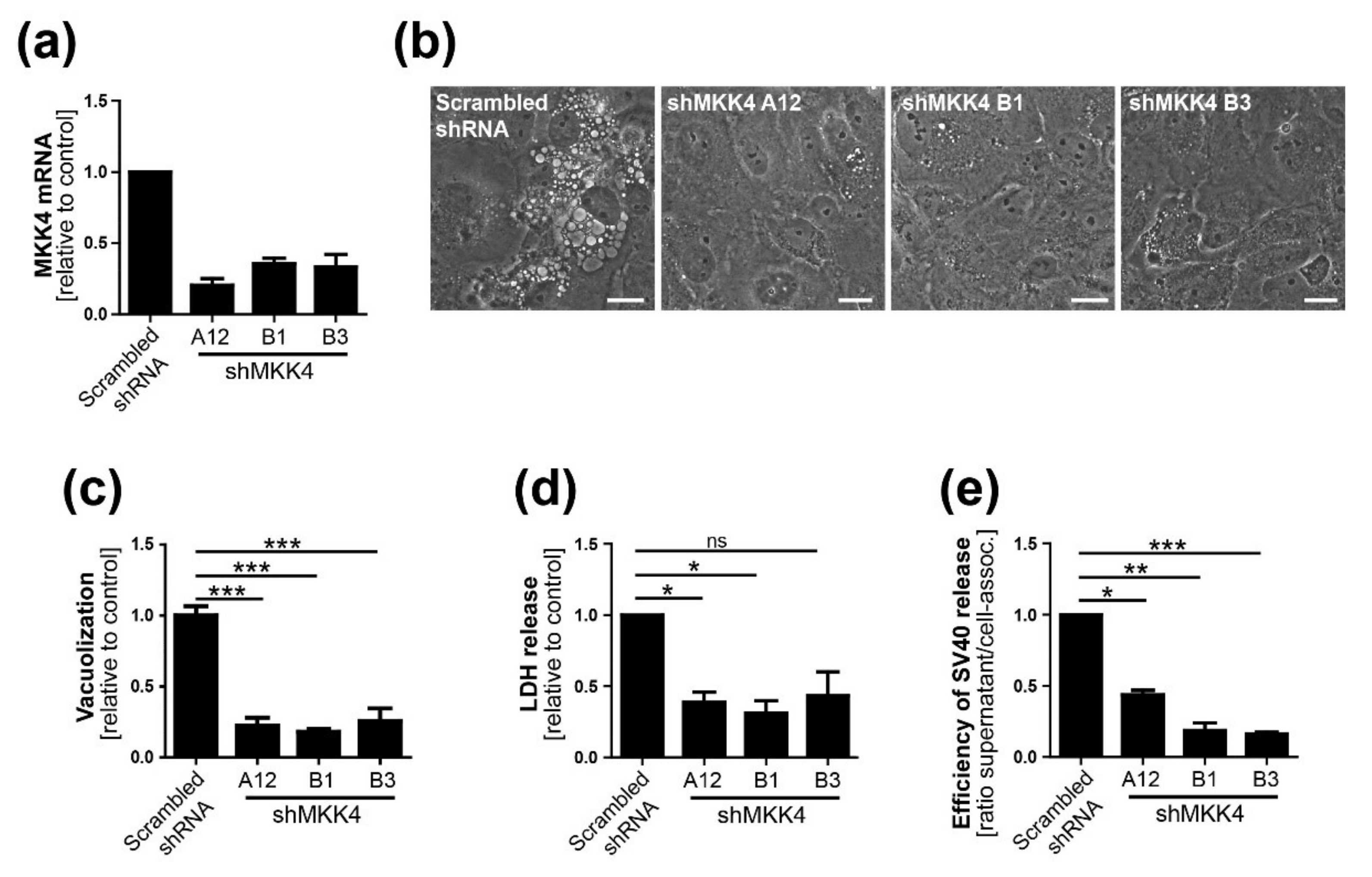
Three different shRNAs to MKK4 were generated using the MISSION library (Sigma, USA) and a lentiviral system consisting of pRSV, pMDL, and pVSV-G. Briefly, the virus was produced by transfection of HEK293 cells with the transfer and packaging vectors using FuGENE6 (Promega, WI, USA). At 24 and 48 h.p.i., the virus-containing supernatant was filtered using a 0.45-µm nylon membrane filter and stored at −80 °C. Pooled virus preparations were used for CV-1 target cell transduction. At about 24 h.p.i., puromycin treatment was started and maintained until the end of experiments. MKK4 knockdown was verified by qPCR.
The following MKK4 shRNAs were used in this study:
A12: TRCN0000001390
(CCGGCTTCTTATGGATTTGGATGTACTCGAGTACATCCAAATCCATAAGAAGTTTTT)
B1: TRCN0000001391
(CCGGGATGTATGAAGAACGTGCCGTCTCGAGACGGCACGTTCTTCATACATCTTTTT)
B3: TRCN0000001393
(CCGGGATATGATGTCCGCTCTGATGCTCGAGCATCAGAGCGGACATCATATCTTTTT)
Scrambled shRNA: SHC002
(CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTT)
2.11. Flow Cytometry
Flow cytometry was done as previously described [
26]. Briefly, to stain for intracellular VP1 and large T antigen, cells were fixed and permeabilized with methanol or 4% PFA/0.5% Triton X-100 and subsequently stained with the primary antibodies PAb 597 and PAb108 against VP1 and large T antigen, respectively. Alexa Fluor 488-labeled donkey anti-mouse antibody (Jackson ImmunoResearch, PA, USA) or goat anti-mouse APC were used as secondary antibodies. Data were acquired on an AccuriC6 or FACS Calibur flow cytometer (BD Biosciences, USA) and analyzed with FlowJo software (Treestar, OR, USA).
2.12. LDH Release Assay
Lactate dehydrogenase (LDH) release into the cell culture supernatant was quantified using the CytoTox 96 nonradioactive cytotoxicity assay (Promega, WI, USA) according to the manufacturer’s instructions. Raw data were collected on a spectrophotometer at 490 nm. Values were calculated as follows: OD (infected sample) − OD (mock-infected sample)/OD (infected biological control) − OD (mock-infected biological control).
2.13. Data Analysis
All data were primarily processed in Microsoft Excel. Statistical analysis and graph production were performed using Prism software (Graphpad, CA, USA). For statistical analysis of data, an unpaired t-test was used.
4. Discussion
Activation of cellular signaling pathways is important during various steps in virus life cycles. For example, virus binding to its surface receptor often triggers signaling cascades that facilitate virus entry and replication. We previously reported that intracellular SV40 VP1 expression alone does not induce vacuolization and that SV40 particles and VP1 are absent from the vacuolar lumen in infected CV-1 cells or in CV-1 cells undergoing vacuolization in response to acute treatment with L1 pentamers [
19]. These findings suggest that vacuolization is the result of a signaling cascade triggered at the cell surface. In this report, we show that, in addition to signaling occurring early in infection, SV40 also induces the MAPK signaling cascade during the late stage of infection when large amounts of VP1 accumulate to support vacuolization and efficient virus release.
Several findings reported here suggest that progeny SV40 particles bind to GM1 at the plasma membrane and trigger GM1-dependent Ras activation and vacuolization to support further virus release. Live-cell microscopy of infected CV-1 cells revealed that SV40-induced vacuoles are positive for endosomal marker proteins (Rab5, Rab7, and LAMP-1) and display a dynamic endocytic nature, resembling LAMP-1-positive cytoplasmic vacuolation induced by Ras activation (HRas GV12) [
25,
29]. We also show that VP1 co-localizes with GM1 and Ras at the limiting membrane of SV40-induced vacuoles arising late in infection. Most importantly, we showed that dominant-negative Ras S17N or inhibition of JNK signaling inhibits vacuole formation. Overall, we hypothesize that, in response to SV40-induced clustering of GM1, Ras is activated and triggers MAPK signaling through Rac1-MKK4-JNK, which results in vacuolization and subsequent cell lysis and virus release. Mechanistic details of how and where VP1 interacts with GM1 to trigger downstream signaling pathways will require additional studies.
Previously published work is consistent with this model. GM1 clustering has been reported to modify active HRas distribution in membrane nanodomains [
30]. Furthermore, overexpression of constitutively active HRas (HRas G12V) leads to extensive vacuolization in glioblastoma cells, with Ras localized at the limiting membrane of cytoplasmic vacuoles [
25]. Like vacuoles formed during SV40 infection, HRas-induced vacuole formation in glioblastoma cells was independent of ERK signaling [
25] and blocked by chemical inhibition of Rac1 [
31]. Furthermore, in both ras-activated glioblastoma cells and SV40-infected cells, vacuoles contain endosomal markers such as Lamp-1 and undergo extracellular fluid uptake resembling macropinocytosis [
24,
25,
32].
Although Ras and VP1 co-localize in vacuolar membranes (
Figure 1C), we have not determined the cellular compartment where GM1-dependent Ras activation occurs during SV40 infection. GM1 can localize to several different membrane compartments including the plasma membrane, Golgi network, and the ER, and the ceramide structure of GM1 influences its trafficking into various compartments [
33]. Similarly, Ras localizes not only to the plasma membrane but also to internal membranes such as the limiting membranes of endosomes, the ER, and the Golgi network [
34]. FRET-based assays demonstrated that EGF stimulation changed the distribution of endogenous active Ras from the plasma membrane to endosomal-like intracellular vesicles, the Golgi network, and the ER [
35]. Moreover, the subcellular localization of active Ras influences signaling through downstream effector pathways [
36]. Thus, SV40 may activate Ras in various cellular compartments at different steps during the viral life cycle, with different biological consequences.
Time course analysis of SV40-induced late cellular signaling revealed that it precedes vacuolization, cell lysis, and virus release. Active phosphorylated forms of JNK were detected 24–36 h.p.i., coincident with VP1 expression, followed by phosphorylation of nuclear transcription factors. Unlike the acute transient signaling induced by virus cell binding or growth factor receptor signaling upon the addition of fresh growth medium, signaling in this late phase of infection is sustained. Activation of cell signaling preceded vacuole formation, with the first vacuoles emerging at 36–48 h.p.i., prior to cell lysis and virus release. Although other cellular kinases such as p38 and ERK were also activated during SV40 infection, inhibitor experiments revealed that only the JNK pathway is essential for vacuolization, cell lysis, and efficient virus release. Importantly, inhibition of JNK, Ras, and MKK4 activity or expression did not interfere with intracellular replication of SV40, so the impaired late events do not merely reflect an early replication block. This is consistent with an earlier published report that MAPK/ERK signaling is not required for early response to SV40 infection [
12] and with a recent genome-wide analysis of kinases that contribute to SV40 endocytosis, which did not detect a requirement for MAPK signaling [
13]. We conclude that Ras-Rac1-MKK4-JNK signaling is essential late during SV40 infection for vacuolization and cell death leading to virus release, although a contribution of other signaling pathway components such as MKK7 cannot be ruled out.
In agreement with previous studies [
37], we showed that infectious SV40 first appears at low levels in the supernatant as early as 36 h.p.i. The release of virus at this early time might occur due to the first cells that lyse or to virus released via a non-lytic process. We hypothesize that SV40 released from cells around this time binds to the plasma membrane of the same and neighboring infected cells and induces cell signaling, which in turn stimulates cell lysis and subsequent increased virus release from these cells, thus establishing a positive feedback loop that stimulates further signaling and virus release. This model is similar to our earlier analysis of vacuolization during SV40 infection [
19], in which we proposed that the first released virus binds to cell surface GM1 and stimulates vacuole formation late during infection. We extend the model here to include virus release as well as vacuolization as a phenotype that can be acutely triggered late in infection by the first progeny virus released. Thus, the initial wave of released virus primes the infected cell population for more pronounced vacuolization and enhanced virus release.
To complete the virus life cycle, polyomavirus release depends on lysis of the infected cell. Whether cell death is the consequence of plasma membrane rupture resulting from extensive virus production or a regulated process depending on active cellular signaling was heretofore unclear. Here, we show that high levels of intracellular virus are not sufficient for efficient SV40 release and that cellular MAPK signaling is necessary for optimal cell lysis and virus release. This lytic process resembles methuosis, which, as noted above, is a Rac1-dependent cell death pathway displaying characteristic cellular vacuolization occurring after ectopic expression of oncogenic Ras [
24,
25,
29].
Vacuolization and virus release are both facilitated by MAPK signaling, and vacuolization precedes virus release. If activation of the Ras-MAPK signaling cascade independently induces both vacuole formation and virus release, vacuolization is a convenient marker for the signaling events that foster efficient release. Alternatively, it is possible that vacuolization itself facilitates subsequent cell death and enhanced virus release.
In addition to the role of cellular signaling, virus-encoded proteins could also be involved in cell lysis and SV40 release. VP4 is a late SV40 protein previously reported to function as a viroporin to support virus release [
38]. VP4 was also reported to disrupt membranes when ectopically added to red blood cells, liposomes, or Cos-7 cells [
38]. However, more recent studies did not confirm the lytic activity of VP4 during SV40 infection [
16]. In the context of SV40 replication, the expression of VP1 alone, without VP2 and VP3, leads to cell lysis and the release of viral particles, suggesting that lytic activity is mediated via VP1, likely through activation of a cellular program as reported here [
14,
16].
Our results raise the possibility that JNK and MAPK pathway inhibitors may have a role in treating polyomavirus infections by decelerating virus propagation and spread within the host by reducing virus release. Such a scenario would presumably provide a protective effect in affected tissues while immune reconstitution is underway [
39]. Because of the long replication cycle of human polyomaviruses and difficulties in synchronizing infection, late events are difficult to study. Nevertheless, further studies of the human pathogenic polyomaviruses may reveal that they are also affected by signaling programs late during infection. Although human polyomaviruses bind only weakly to GM1 [
39], a variety of stimuli can activate the signaling elements described here. Thus, analysis of late events of human polyomavirus infection might establish that this or overlapping signaling pathways are viable therapeutic targets.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







