The Autophagy Protein ATG16L1 Is Required for Sindbis Virus-Induced eIF2α Phosphorylation and Stress Granule Formation


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Antibodies for Western Blotting and Immunostaining
2.3. Immunoblotting
2.4. Immunocytochemistry and Fluorescent Microscopy
2.5. Reverse Transcription PCR
2.6. Statistical Analysis
3. Results
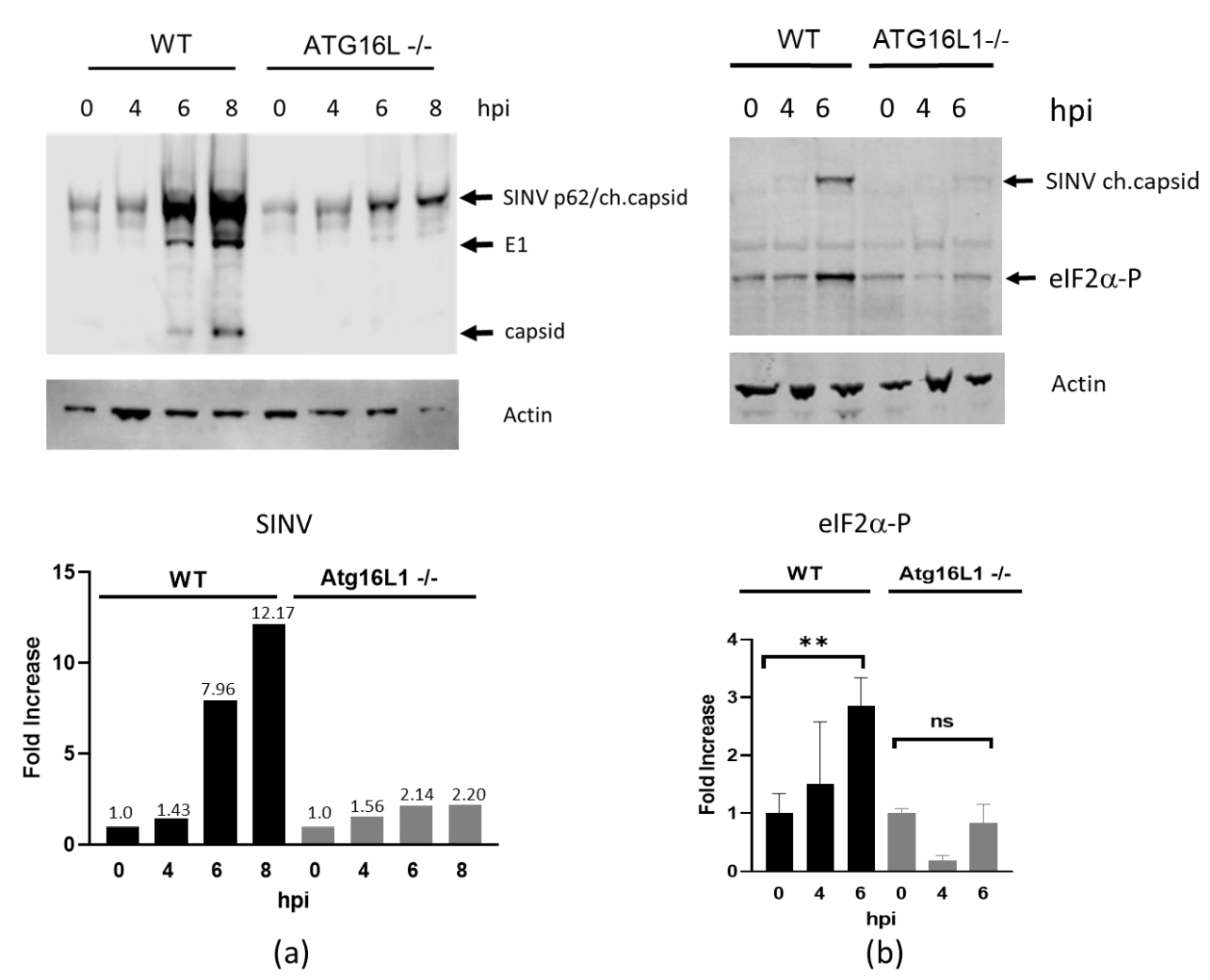
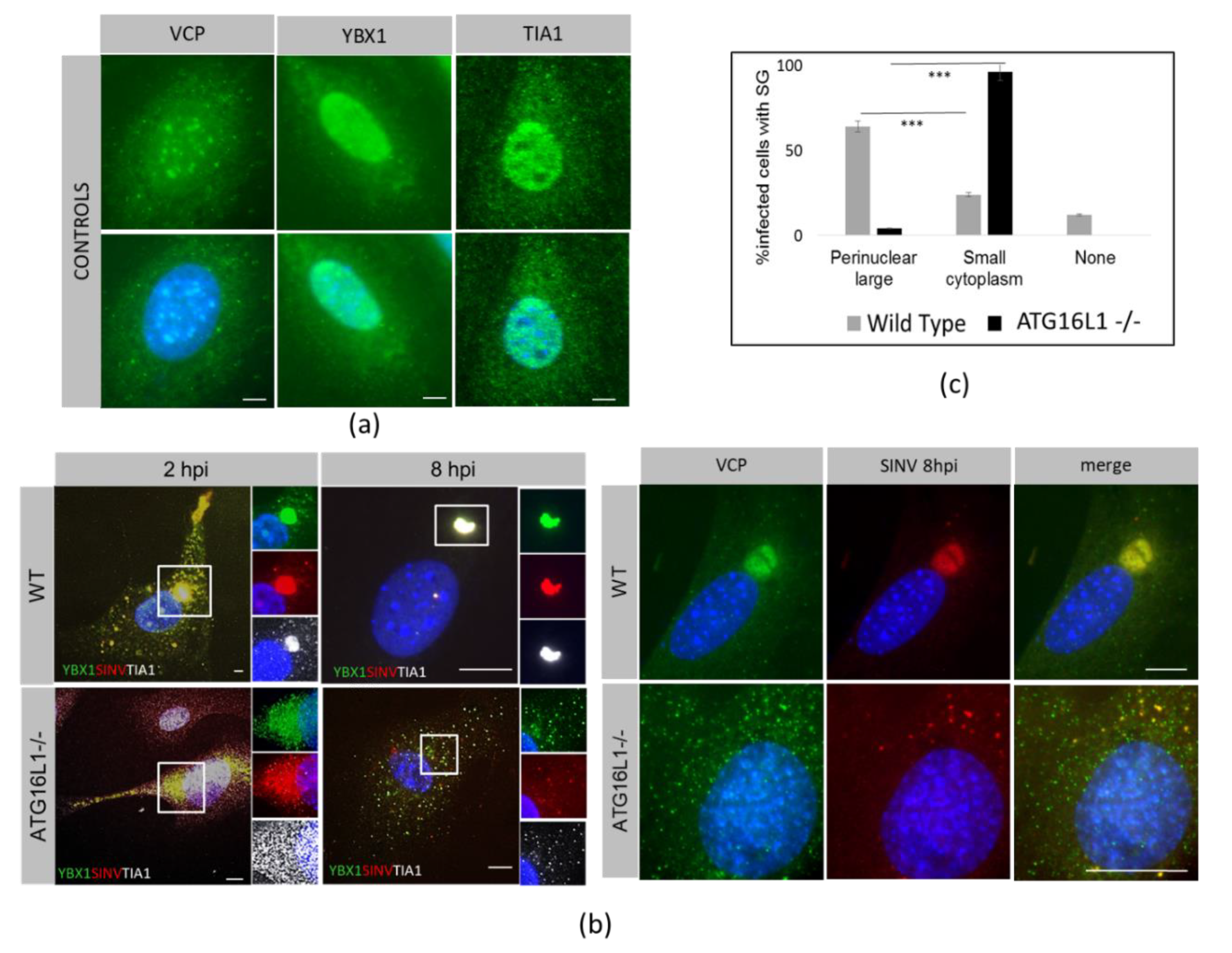
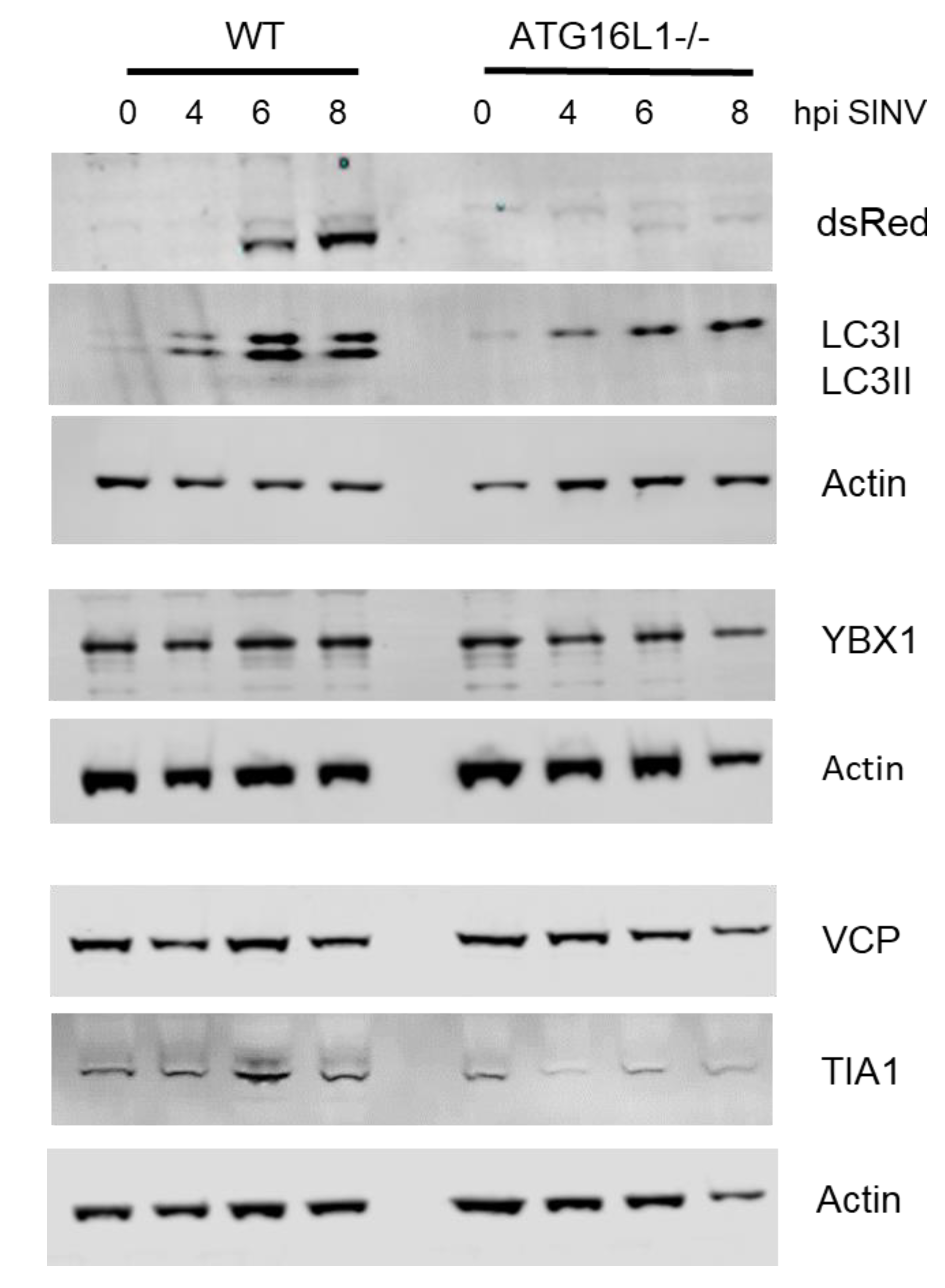
3.1. ATG16L1 is Required for SINV-Induced Phosphorylation of eIF2α and the Switch to Viral Protein Synthesis
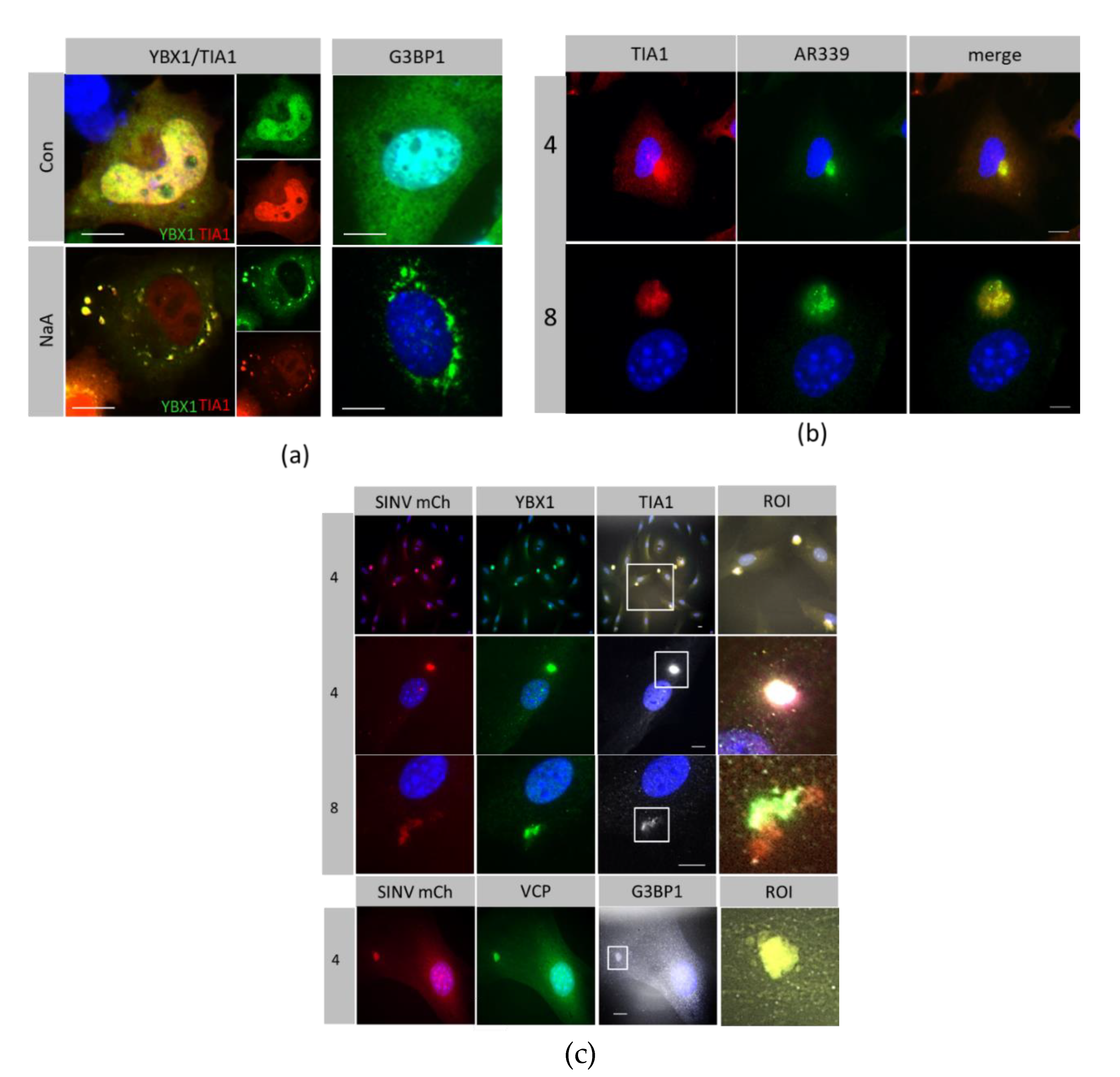
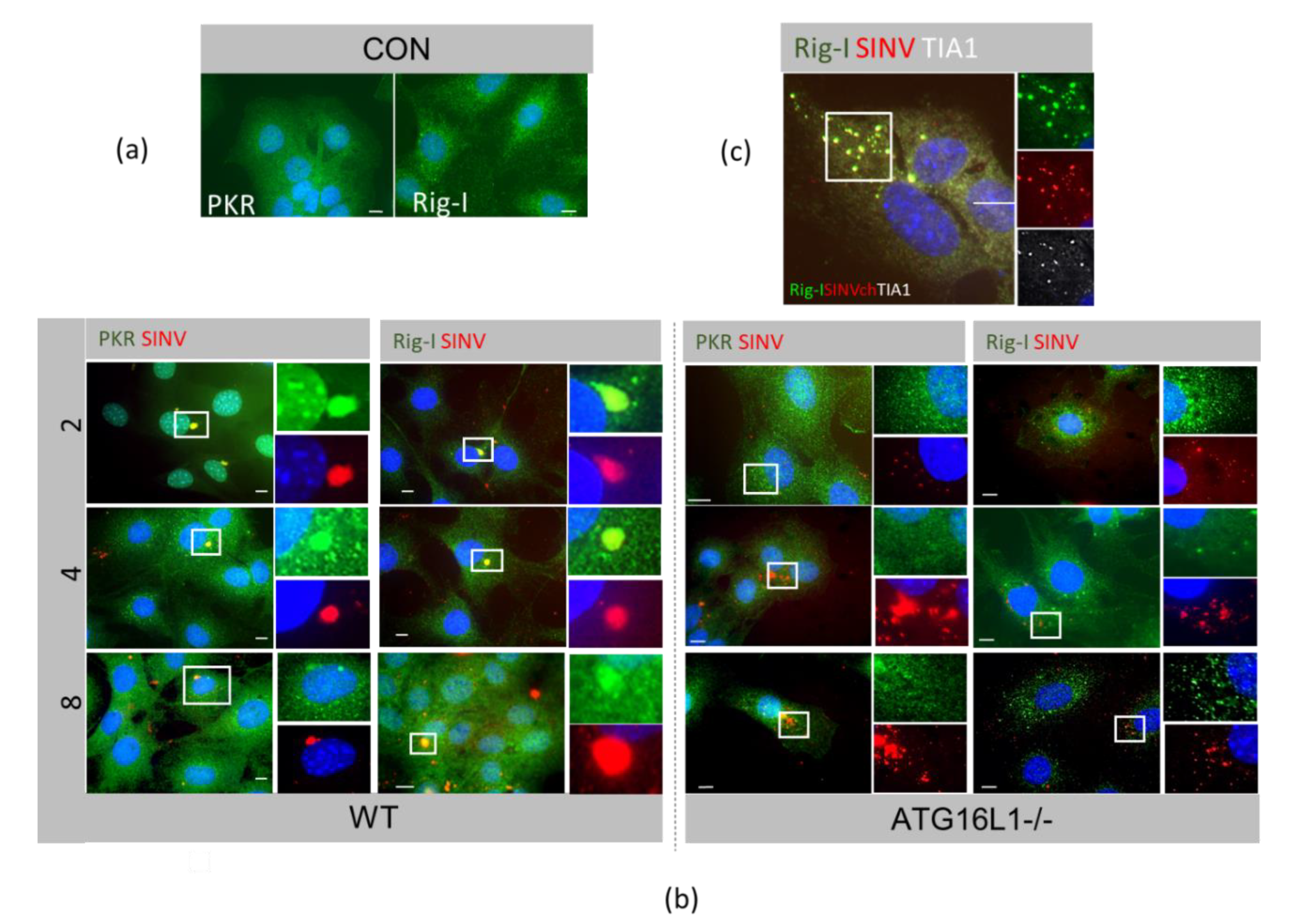
3.2. Host Protein Rearrangements Following Entry of SINV Capsids
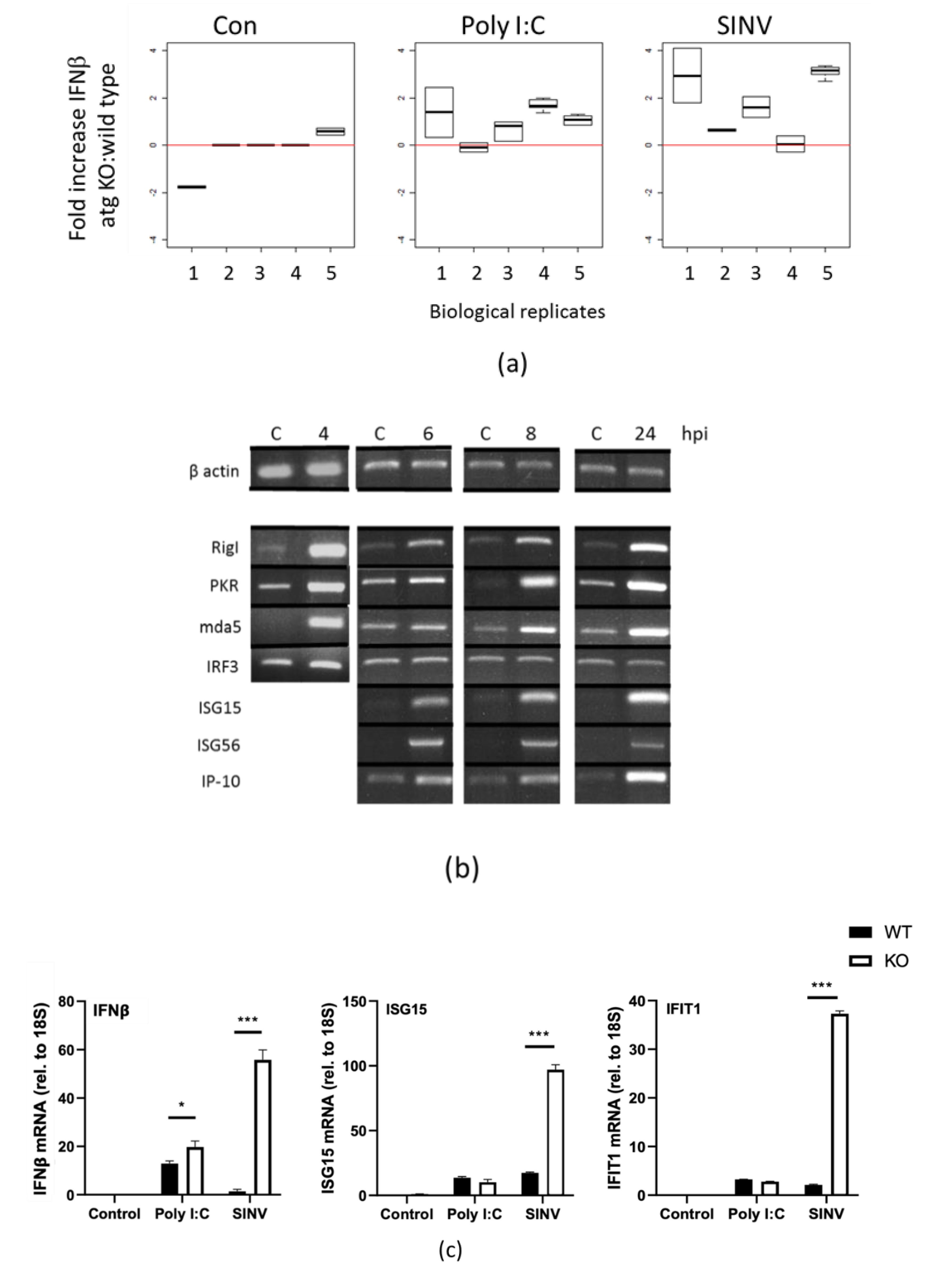
3.3. Innate Responses to SINV in Autophagy-Deficient Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–7372. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Lloyd, R.E. Cytoplasmic RNA Granules and Viral Infection. Ann. Rev. Virol. 2014, 1, 147–170. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Chattopadhyay, S.; Sen, G.C. No Love Lost Between Viruses and Interferons. Ann. Rev. Virol. 2015, 2, 549–572. [Google Scholar] [CrossRef] [PubMed]
- Reineke, L.C.; Kedersha, N.; Langereis, M.; van Kuppeveld, F.J.; Lloyd, R.E. Stress granules regulate double-stranded RNA-dependent protein kinase activation through a complex containing G3BP1 and Caprin1. mBio 2015, 6, e02486. [Google Scholar] [CrossRef] [PubMed]
- Protter, D.S.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef]
- Orvedahl, A.; Levine, B. Autophagy in Mammalian antiviral immunity. Curr. Top. Microbiol. Immunol. 2009, 335, 267–285. [Google Scholar]
- Rupp, J.C.; Sokoloski, K.J.; Gebhart, N.N.; Hardy, R.W. Alphavirus RNA synthesis and non-structural protein functions. J. Gen. Virol. 2015, 96, 2483–2500. [Google Scholar] [CrossRef]
- Eng, K.E.; Panas, M.D.; Murphy, D.; Karlsson Hedestam, G.B.; McInerney, G.M. Accumulation of autophagosomes in Semliki Forest virus-infected cells is dependent on expression of the viral glycoproteins. J. Virol. 2012, 86, 5674–5685. [Google Scholar] [CrossRef]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Tallóczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010, 7, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Wileman, T. Autophagy as a defense against intracellular pathogen. Essays Biochem. 2013, 55, 153–163. [Google Scholar] [PubMed]
- Rai, S.; Arasteh, M.; Jefferson, M.; Pearson, T.; Wang, Y.; Zhang, W.; Bicsak, B.; Divekar, D.; Powell, P.P.; Naumann, R.; et al. The ATG5-binding and coiled coil domains of ATG16L1 maintain autophagy and tissue homeostasis in mice independently of the WD domain required for LC3-associated phagocytosis. Autophagy 2019, 15, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; MacDonald, M.R.; Rice, C.M. To translate, or not to translate: Viral and host mRNA regulation by interferon-stimulated genes. Trends Cell Biol. 2015, 25, 320–329. [Google Scholar] [CrossRef]
- Fros, J.J.; Pijlman, G.P. Alphavirus Infection: Host Cell Shut-Off and Inhibition of Antiviral Responses. Viruses 2016, 8, 166. [Google Scholar] [CrossRef]
- Berlanga, J.J.; Ventoso, I.; Harding, H.P.; Deng, J.; Ron, D.; Sonenberg, N.; Carrasco, L.; de Haro, C. Antiviral effect of the mammalian translation initiation factor 2 alpha kinase GCN2 against RNA viruses. EMBO J. 2006, 25, 1730–1734. [Google Scholar] [CrossRef]
- Ventoso, I.; Sanz, M.A.; Molina, S.; Berlanga, J.J.; Carrasco, L.; Esteban, M. Translational resistance of late alphavirus mRNA to eIF2alpha phosphorylation: A strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev. 2006, 20, 87–100. [Google Scholar] [CrossRef]
- Frolov, I.; Schlesinger, S. Translation of Sindbis virus mRNA: Analysis of sequences downstream of the initiating AUG codon that enhance translation. J. Virol. 1996, 70, 1182–1190. [Google Scholar]
- McCormick, C.; Khaperskyy, D.A. Translation inhibition and stress granules in the antiviral immune response. Nat. Rev. Immunol. 2017, 17, 647–660. [Google Scholar] [CrossRef]
- Tallóczy, Z.; Jiang, W.; Virgin, H.W., 4th; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 2, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.M.J.; Mellouk, N.; Brumell, J.H. An autophagy-independent role for ATG16L1: Promoting lysosome-mediated plasma membrane repair. Autophagy 2019, 5, 932–933. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef] [PubMed]
- Onomoto, K.; Yoneyama, M.; Fung, G.; Kato, H.; Fujita, T. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014, 35, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Akira, S. Regulation of innate immune responses by autophagy-related proteins. J. Cell Biol. 2010, 189, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.L.; Gardner, C.L.; Kimura, T.; White, J.P.; Liu, G.; Trobaugh, D.W.; Huang, C.; Tonelli, M.; Paessler, S.; Takeda, K.; et al. A viral RNA structural element alters host recognition of nonself RNA. Science 2014, 343, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Gorchakov, R.; Frolova, E.; Williams, B.R.; Rice, C.M.; Frolov, I. PKR-dependent and –independent mechanisms are involved in translational shutoff during Sindbis virus infection. J. Virol. 2004, 78, 8455–8467. [Google Scholar] [CrossRef]
- Atasheva, S.; Kim, D.Y.; Frolova, E.I.; Frolov, I. Venezuelan equine encephalitis virus variants lacking transcription inhibitory functions demonstrate highly attenuated phenotype. J. Virol. 2015, 89, 71–82. [Google Scholar] [CrossRef]
- Seguin, S.J.; Morelli, F.F.; Vinet, J.; Amore, D.; De Biasi, S.; Poletti, A.; Rubinsztein, D.C.; Carra, S. Inhibition of autophagy, lysosome and VCP function impairs stress granule assembly. Cell Death Differ. 2014, 21, 1838–1851. [Google Scholar] [CrossRef]
- Onomoto, K.; Jogi, M.; Yoo, J.S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS ONE 2012, 7, e43031. [Google Scholar] [CrossRef]
- Panas, M.D.; Varjak, M.; Lulla, A.; Eng, K.E.; Merits, A.; Karlsson Hedestam, G.B.; McInerney, G.M. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol. Biol. Cell 2012, 23, 4701–4712. [Google Scholar] [CrossRef] [PubMed]
- Gorchakov, R.; Garmashova, N.; Frolova, E.; Frolov, I. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J. Virol. 2008, 82, 10088–10101. [Google Scholar] [CrossRef] [PubMed]
- Scholte, F.E.; Tas, A.; Albulescu, I.C.; Žusinaite, E.; Merits, A.; Snijder, E.J.; van Hemert, M.J. Stress granule components G3BP1 and G3BP2 play a proviral role early in Chikungunya virus replication. J. Virol. 2015, 89, 4457–4469. [Google Scholar] [CrossRef] [PubMed]
- Fros, J.J.; Domeradzka, N.E.; Baggen, J.; Geertsema, C.; Flipse, J.; Vlak, J.M.; Pijlman, G.P. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J. Virol. 2012, 86, 10873–10879. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jefferson, M.; Bone, B.; Buck, J.L.; Powell, P.P. The Autophagy Protein ATG16L1 Is Required for Sindbis Virus-Induced eIF2α Phosphorylation and Stress Granule Formation. Viruses 2020, 12, 39. https://doi.org/10.3390/v12010039
Jefferson M, Bone B, Buck JL, Powell PP. The Autophagy Protein ATG16L1 Is Required for Sindbis Virus-Induced eIF2α Phosphorylation and Stress Granule Formation. Viruses. 2020; 12(1):39. https://doi.org/10.3390/v12010039
Chicago/Turabian StyleJefferson, Matthew, Benjamin Bone, Jasmine L. Buck, and Penny P. Powell. 2020. "The Autophagy Protein ATG16L1 Is Required for Sindbis Virus-Induced eIF2α Phosphorylation and Stress Granule Formation" Viruses 12, no. 1: 39. https://doi.org/10.3390/v12010039
APA StyleJefferson, M., Bone, B., Buck, J. L., & Powell, P. P. (2020). The Autophagy Protein ATG16L1 Is Required for Sindbis Virus-Induced eIF2α Phosphorylation and Stress Granule Formation. Viruses, 12(1), 39. https://doi.org/10.3390/v12010039