1. Introduction
The cervid farming industry is a fast growing industry in rural Florida and other parts of North America [
1]. This venture is significant in scope because the industry generated almost US
$8 billion for the U.S. economy and employed > 56,000 people by the year 2017 [
1]. In the United States, cervid farming includes reindeer (
Rangifer tarandus) in Alaska, elk (
Cervus elaphus) in the Midwest, and white-tailed deer (
Odocoileus virginianus) in many states such as Florida, Pennsylvania, and Texas. White-tailed deer are the most commonly farmed deer species in the continental United States [
2]. Deer farming has positive impacts on local economies, and game preserves have the potential to conserve wild lands that might otherwise be developed commercially; however diseases can be passed between wild and farmed populations with potential impacts to both populations. As in other livestock systems, the spread of infectious diseases among wild and farmed deer can occur through arthropod vector-borne transmission routes, via inhalation of aerosolized virus particles, through nose-nose contact between farmed and wild deer, or through contact with fomites that can be wind-borne or spread through naturally occurring disasters. Appropriate biosecurity measures can reduce the risk of transmission of many pathogens, and therefore reduce the economic and biological impacts of disease outbreaks. These epizootic events can impact local economies as in the case of chronic wasting disease, and spillover transmission can impact wildlife communities [
3,
4].
Bluetongue virus (BTV) and epizootic hemorrhagic disease virus (EHDV) are orbiviruses that are transmitted by
Culicoides spp., and these viruses can cause high morbidity and mortality in farmed and wild populations of white-tailed deer in North America [
4,
5]. Of note, both viruses also affect other wildlife species including mule deer (
Odocoileus hemionus), pronghorn (
Antilocapra americana), elk, mountain goat (
Oreamnos americanus), and bighorn sheep (
Ovis canadensis) [
6], as well as domestic livestock such as cattle [
7]. Both viruses circulate in Florida where they are considered endemic in wild populations [
5,
8,
9,
10,
11], yet cause considerable production losses in farmed populations. Another reovirus, i.e., mammalian orthoreovirus type 2, has recently been reported to be associated with mortality of farmed white-tailed deer fawn in Florida, likely exemplifying yet another important reovirus pathogen of farmed white-tailed deer populations [
12].
Despite the economic hardship these reoviruses impose on Florida farmed deer, the viral diseases of farmed deer in Florida and elsewhere are underexplored, and thus a better understanding of (1) the potential infectious diseases that may arise in deer farms, and (2) effective methods to curtail or prevent these diseases, are essential for economic profitability and to minimize the risk of spreading infectious diseases to susceptible wildlife and farmed animals [
4]. Identification of the reoviruses in circulation among cervids is essential for the production of preventive vaccines, and for those that are arthropod-borne, control of their vectors.
Viruses of the family
Reoviridae (“reoviruses”) are non-enveloped, icosahedral viruses that have a triple-capsid structure, and may appear spherical in shape. There are 15 genera of reoviruses, and these are divided into two subfamilies:
Spinareovirinae, which have relatively large spikes or turrets situated at the 12 icosahedral vertices of either the virion or core particle, and
Sedoreovirinae, which do not have large surface projections on their virions or core particles, giving them an almost spherical or “smooth” appearance. The genus
Orbivirus is 1 of the 15 genera within the family
Reoviridae, and belongs in the subfamily
Sedoreovirinae. Currently, there are 22 recognized
Orbivirus species (representing 22 distinct virus serogroups) that have been recognized by the International Committee for the Taxonomy of Viruses (ICTV), and at least 10 unclassified or unassigned viruses [
13].
Orbivirus genomes consist of 10 to 12 dsRNA segments that encode seven major structural proteins (virion proteins, VP1–VP7) and 3 to 5 major non-structural proteins (NS1-NS5); one copy of each segment is packaged per virion [
13,
14,
15]. The virus genomic RNA (vgRNA) segments are mostly monocistronic; a few have two or three in-frame initiation codons that lead to expression of additional open-reading frames (ORFs), apparently through leaky scanning [
13]. In addition, at least for BTV, a frame-shift initiation codon in one of the genomic segments leads to production of NS4 and Vp6 (helicase). The 10 to 12 genomic segments (Seg) occur in three distinct size classes based on their molecular weight (MW), and these are readily identified through their electrophoretic mobility (i.e., by their electropherogram patterns). These segments are classified as large (L1, L2, and L3), medium (M1, M2, and M3), and small (S1, S2, S3 and S4) and in order of decreasing MW as Seg-1 to Seg-10. The orbivirus
RNA-dependent RNA polymerase (
RdRp) gene is encoded by Seg-1, the protein is traditionally termed virus protein 1 (VP1), and its gene traditionally referred to as the
VP1 instead of
RdRp gene. The amino acid (aa) sequence of VP1 (RdRp protein) is an important marker for orbivirus classification. An aa identity > 30% with that of other characterized orbiviruses is required to be a member of the genus
Orbivirus. Available data suggests that isolates from different genera usually have < 26% aa identity in comparisons between their RdRps, while within a single genus identities are usually > 33% [
13,
14]. Of note, the terminology for orbivirus genome segment numeration and protein designation has not been uniform; some laboratories classify orbivirus proteins according to the size of the proteins, whereas others use the size of genome segments from which they are encoded [
16]. In this manuscript, for each vgRNA electropherogram position, the protein encoded has been identified in silico, and thus correlates with the size of its coding segment. The aa sequence of the sub-core protein (T2 protein, traditionally referred to as VP3) is encoded by Seg-2, and is used to classify species within genera. Orbiviruses within a single species group show greater than 91% identity of their T2 aa sequences [
13]. Seg-3 encodes the major outer capsid protein (VP2), and viruses within the same species have > 76% nucleotide identity while those in different species usually have < 74% identity, and these differences are also reflected in the amino acid sequences of the viral proteins [
13]. Of note, VP2 is the major determinant of serotype, with a minor role for VP5 [
13,
17,
18].
Orbiviruses occur globally and infect a wide variety of vertebrate hosts, including bats, birds, marsupials, rodents, sloths, human and non-human primates, and wild and domestic ruminants and equids. They are common in insects and ticks, where their pathogen status is under-explored and thus not clear, but select viruses can cause diseases in vertebrates including humans. Unlike the reoviruses that replicate only in vertebrate species and are transmitted between hosts by respiratory or fecal–oral routes, orbiviruses replicate in both hematophagous vectors (
Culicoides biting midges, anopheline or culicine mosquitoes, phlebotomine sandflies, and ticks, depending on the virus) and vertebrate hosts. They replicate in and are transmitted between their vertebrate and arthropod vectors. Some orbiviruses such as African horse sickness virus, BTV, EHDV, and Peruvian horse sickness virus (PHSV) are considered serious animal pathogens, while a few such as the Kemerovo, Lebombo, Orungo, and Tribeč viruses are considered “moderate” human pathogens [
19].
The University of Florida (UF) Cervidae Health Research Initiative (CHeRI) is a multidisciplinary entity focused on improving the health and production of captive and farmed cervids of Florida. One research focus is on the identification of viruses that impact the health of Florida’s farmed deer. Florida deer farmers can access free diagnostic assays by contacting CHeRI when they have a deceased deer on their farm. Deer farmers have two options, to collect and send samples to the CHeRI lab or ask a technician to perform a field necropsy of the animal. Upon request, the CHeRI also assists with the identification of deer viruses affecting farmed deer outside of Florida.
We report the detection and characterization of three novel orbivirus species from farmed white-tailed deer that were found dead in their pens. To our knowledge, none of these viruses have previously been reported in North America.
2. Materials and Methods
2.1. Field Collection of Deer Organ and Tissue Specimens
CHeRI provides guidelines on its website for the collection of organs or tissue specimens from dead deer and their shipment to UF for diagnostic pathology work:
http://www.wec.ufl.edu/CHeRI/diagnostics/. A non-technical movie is also available:
https://www.youtube.com/watch?v=83UKkeXYWik&feature=youtu.be. As possible (depending on specimen integrity and type, and storage procedures), histopathology, microbiology, and virology analyses are performed. Briefly, during field necropsy and gross examination, diseased portions of organs (typically heart, kidney, liver, lungs, and the spleen) are collected as soon as possible after an animal’s death and placed into sterile polyethylene Whirl-Pak
TM bags (Nasco, Ft. Atkinson, WI, USA). When possible, whole peripheral blood is collected from recently deceased animals and injected into EDTA blood collection tubes (K
2EDTA tubes, Becton, Dickinson, and Company, Franklin Lakes, New Jersey, USA). Blood is used for initial RT-PCR screens for the presence of BTV and EHDV vgRNAs, as: (1) BTV and EHDV bind to and remain associated with red blood cells, and (2) RNA purification from blood is relatively straightforward using a variety of commercial RNA purification kits, and thus RT-PCR tests for BTV and EHDV vgRNAs can be promptly initiated from blood specimens for rapid reporting of results to deer farmers. Because of post-mortem peripheral blood coagulation, cardiac blood is collected instead and analyzed for blood-borne pathogens from animals dead for more than two hours. In that instance, an aliquot of cardiac blood is obtained by syringe then injected into EDTA blood collection tubes. After collection, anticoagulated blood (in EDTA blood collection tubes) is maintained refrigerated including during shipment to CHeRI for virology tests. If the RT-PCR tests are negative for BTV and EHDV vgRNAs, the tests are repeated using RNA extracted from spleen tissue. Tissue and organ samples recovered from newly deceased animals are kept refrigerated for histopathology analyses, and microbiology (bacteriology and mycology) and virology work-up. If decomposition of tissues is evident, tissue and organ specimens are transported on cool packs to the UF for microbiology (bacteriology and mycology) and virology work-up only. For virus isolation, emphasis to date at the CHeRI-affiliated laboratories has been on the detection and isolation of BTV and EHDV, as to date, they have been the most common agents of virus-induced mortality in the farmed deer of Florida. Based on our cumulative experiences, both EHDV and BTV can be readily isolated from spleen tissues collected within 48 h of death of cervids that have died of hemorrhagic disease; we have not attempted to isolate the viruses from spleen tissues collected at later time points. All work was approved by the Institutional Animal Care and Use Committee at the University of Florida (IACUC Protocol Numbers 201609390, initiated 24 May 2016, and 201909390, initiated 21 March 2019).
2.2. White-Tailed Deer History and Specimens Collected for Tests
Background information regarding each animal is provided below; the prefix “OV” for each animal identification number denotes Odocoileus virginianus.
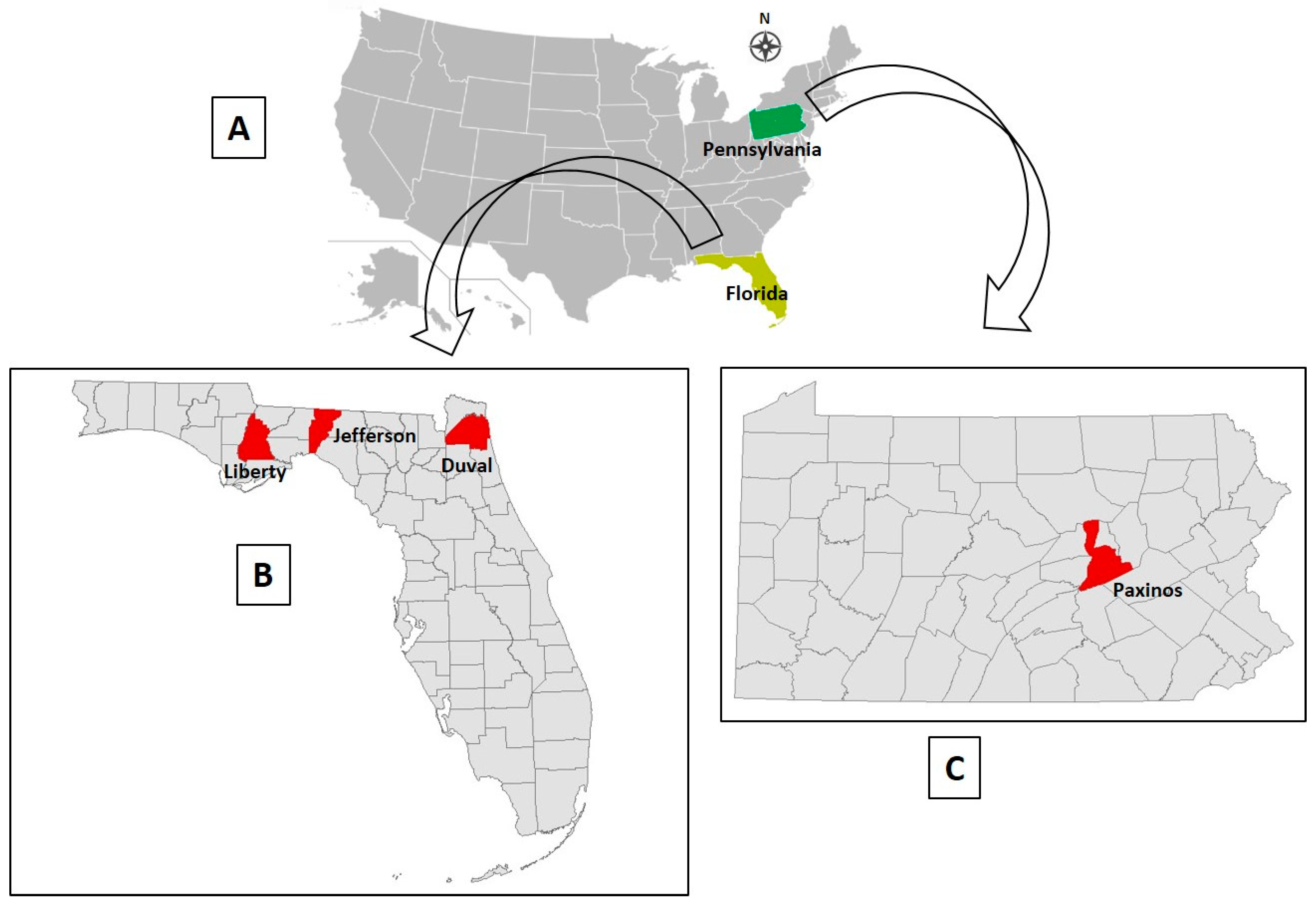
OV610. On August 30, 2017, a farmed 2-year old buck in Paxinos County, Pennsylvania (
Figure 1), was found dead in its pen. The farm’s owner noted it was healthy, appearing normal the previous day. A field necropsy was performed by a veterinarian on August 31, 2017, whereupon heart, intestine, liver, lung, kidney, skeletal muscle, spleen, ocular fluid, and rumen contents were collected and sent to the CHeRI for possible diagnostic microbiology (culture), histopathology, and virology evaluations. The spleen was used for virus isolation attempts, as either BTV or EHDV was suspected to be the causative agent, and as noted in
Section 2.1 above, either virus can be isolated from a spleen collected < 48 h post-mortem.
OV617. On September 10, 2017, a 3-month old fawn (sex not specified by farmer) in Jefferson County, northwest Florida (
Figure 1), died. The animal was observed to be ill the previous day, when it was administered a dose of Draxxin (tulathromycin), a triamilide antibiotic indicated for the treatment of bovine and swine bacterial respiratory diseases and bovine foot-rot. Draxxin is also used in deer farming to help control bacterial infections of the lung, as pneumonia is a common consequence of EHDV infection. On September 11, postmortem heart, liver, lung, kidney, and spleen tissues were collected by the deer farm owner and placed into sterile Whirl-Pak bags and an aliquot of venous blood was collected. All the samples were maintained refrigerated until arrival at CHeRI on September 14, 2017. As the organ and tissue samples were collected > 24 h post-mortem and were decomposing, none were submitted for histopathology or microbiology analyses, though as previously noted, spleen tissue was used for virus isolation attempts as it had been collected < 48 h post-mortem.
OV682. On November 24, 2017, a previously healthy farmed 5-year old doe in Jefferson County, Florida (
Figure 1), was found recumbent with its ears down and did not allow anyone to approach her. Later that day, the animal was unable to rise, unresponsive to stimuli, and died. A field necropsy was performed the following day by the deer farm owner, nearly 24 h after the animal had died, whereupon cardiac blood, heart, kidney, liver, lung, spleen, and small intestine tissues were removed and maintained refrigerated until delivery to CHeRI on November 30, 2017. Since necropsy was performed nearly 24 h post-mortem and the stage of decomposition somewhat advanced, no tissues were submitted for histopathology work. Lung tissue was submitted for diagnostic microbiology analyses, and spleen tissue was used for virus isolation attempts as it had been collected < 48 h post-mortem.
OV862. On September 18, 2018, a previously healthy farmed 1-year-old buck was found dead in Liberty County, FL (
Figure 1). A necropsy was performed the same day by a CHeRI technician. Cardiac blood, and heart, kidney, liver, lung and spleen tissues were collected. As only the lung and kidney tissues appeared diseased, those were the only tissues submitted to the CHeRI for microbiology analyses. Spleen tissue was used for virus isolation attempts.
OV867. On September 20, 2018, a previously healthy farmed 3-month-old doe fawn in Duval County, Florida (
Figure 1), was found struggling in the early morning, then died mid-morning. No signs of diseases had been observed the prior day. A necropsy was performed a few hours post-mortem by the farm owner, who only collected a kidney for microbiology analyses, and spleen tissue for virus isolation attempts.
OV926. On December 15, 2018, a previously healthy farmed 1-year-old white-tailed deer doe was found dead in Liberty County, Florida (
Figure 1). No medications had been administered to the animal within the 3 months prior to death. A necropsy was performed on December 17, 2018, when the animal was at an advanced state of decomposition, and this precluded collection of tissues for diagnostic microbiology and histopathology. Spleen tissue was collected for virus isolation attempts.
2.3. Extraction of vgRNA from Virions in Blood and Tissue Homogenates for Initial RT-PCR Screens
Virus genomic RNA was extracted from virions in blood and tissue homogenates using a QIAamp Viral RNA mini kit (Qiagen, Valencia, California, USA) following the manufacturer’s protocol. SUPERase-In RNase inhibitor (Thermo Fisher Scientific, Waltham, Massachusetts, USA, Cat. Number AM2694) was added to the purified RNA samples, which were immediately stored at –80 °C to preserve them for molecular tests at a later time.
2.4. RT-PCR Screens for EHDV and BTV vgRNAs
RT-PCR screens for EHDV and BTV vgRNAs were performed following the protocol described by Ahasan, et al. [
12]. Briefly, the RT-PCR multiplex assay for five viruses described by Wernike et al. [
20] was modified for detection of BTV and EHDV vgRNAs only. Each individual RT-PCR reaction was performed in a total reaction volume of 25 μL using a VetMAX-Plus Multiplex One Step RT-PCR kit (Applied Biosystems, Foster City, California, USA). Individual reactions contained 12.5 μL 2X RT-PCR buffer, 2.5 μL 10X multiplex RT-PCR enzyme mix, 1 μL Xeno VIC Assay (which contains proprietary forward and reverse primers and a 5′ 2′-chloro-7′-phenyl-1,4-dichloro-6-carboxyfluorescein (VIC) flurophore and 3′ MGB (minor grove binder) NFQ (nonfluorescent quencher)–labeled probe, specific for an internal control RNA supplied with the kit), 0.1 μL of the Xeno RNA (internal control) (Applied Biosystems), 10 pmol of BTV forward and reverse primers, 2 pmol of BTV probe (fluorophore: 6-Carboxyfluorescein (6-FAM); quencher: Black Hole Quencher-1 (BHQ1), 15 pmol of EHDV forward and reverse primers, 2.5 pmol of EHDV probe (flurophore Texas Red; quencher BHQ2), and RNAse-free water to yield 20 μL master mix. After addition of 5 μL of template RNA, RT-PCR was performed in a 7500 fast Real-Time PCR System with the following thermal profile: reverse transcription step at 48 °C for 10 min, initial denaturation step at 95 °C for 10 min followed by 40 cycles of 3-step cycling consisting of denaturation at 95 °C for 15 s, annealing at 57 °C for 45 s, and final extension at 68 °C for 45 s.
2.5. RT-PCR Tests for Eastern Equine Encephalitis Virus (EEEV) and West Nile Virus (WNV) vgRNAs
RNA purified from spleen homogenate was also tested for EEEV and WNV vgRNAs, as the viruses are endemic in Florida and are known cervid pathogens. Both assays were carried out using the VetMAX Plus One Step RT-PCR kit (Applied Biosystems). Individual 25-μL reactions contained 12.5 μL 2X RT-PCR Buffer, 1 μL 25X RT-PCR enzyme, RNAase-free water, 1 μL Xeno VIC assay primers and probe, and 0.1 μL Xeno RNA (internal control), to which was added: (1) for WNV, 25 pmol of each primer and 5 pmol of probe: fluophore 6-FAM, quencher BHQ1, based on the procedure of Lanciotti et al. [
21], or (2), for EEEV, 18 pmol of each primer and 5 pmol of probe: flurophore 6-FAM, quencher BHQ1, based on the procedure of Lambert et al. [
22], yielding a 21-µL master-mix. Thereafter, 4 μL of template RNA was added to 21 μL of the master mix containing either WNV or EEEV specific primers and probe, and RT-PCRs performed in a 7500 fast Real-Time PCR System as follows: (1) for WNV, reverse transcription at 48 °C for 10 min, initial denaturation step at 95 °C for 10 min followed by 40 cycles of 2-step cycling consisting of 95 °C for 15 s and annealing/extension at 60 °C for 45 s, and (2) for EEEV, reverse transcription step at 48 °C for 10 min, initial denaturation step at 95 °C for 10 min followed by 40 cycles of 2-step cycling consisting of denaturation at 95 °C for 15 s and annealing/extension at 60 °C for 45 s.
2.6. RT-PCR Typing of EHDV
Presumptive determination of EHDV type was accomplished using RNA purified from spleen tissue following the method of Maan et al. [
23]. Tests were performed with Seg-2 primers for discrimination between EHDV types 1, 2, or 6 only, as these are the typical EHDV types encountered in Florida and the rest of the United States. Each individual RT-PCR reaction was performed in a total reaction volume of 25 μL using a VetMAX-Plus Multiplex One Step RT-PCR kit (Applied Biosystems). Individual reactions contained 12.5 μL 2X RT-PCR buffer, 1.0 μL 25X multiplex RT-PCR enzyme mix, 1 μL Xeno VIC Assay primers and probe, and 0.1 μL Xeno RNA (internal control) (Applied Biosystems). For EHDV-1, each RT-PCR reaction contained 10 pmol of EHDV-1w/275F primer, EHDV-1w/341R primer, and 2.5 pmol of EHDV-1w/329P.probe, Similarly, for EHDV-2, each RT-PCR reaction contained 10 pmol each of EHDV-2w/1934F and EHDV-2w/2069R primers, and 2.5 pmol of EHDV-2w/1997P probe, and for EHDV-6, each RT-PCR reaction contained 10 pmol each of EHDV-6e/622F and EHDV-6e/724R primers and 2.5 pmol of EHDV-6e/669P probe. Probes for EHDV-1, -2, and -6 were 5′ labeled with 6-FAM flurophore and the quencher was BHQ1. To each EHDV-1, -2, and -6 reaction mixture, RNAse-free water was subsequently added to yield 20-μL master mixes. After addition of 5 μL of template RNA, RT-PCR was performed in a 7500 fast Real-Time PCR System with the following thermal profile: reverse transcription step at 50 °C for 10 min, initial denaturation step at 95 °C for 10 min followed by 50 cycles of 2-step cycling consisting of denaturation at 95 °C for 30 s and 60 °C degrees for 60 s.
2.7. Propagation of Cell Cultures for Virus Isolation
Cell lines used for virus isolation were obtained from the American Type Culture Collection (ATCC, Manassas, Virginia, USA). As BTV and EHDV were assumed to be the main agents of hemorrhagic disease in Florida’s farmed deer, virus isolation was attempted in two cell lines that are usually susceptible to and permissive for the viruses: C6/36 (
Aedes albopictus (mosquito), ATCC CRL1660) and Vero E6 (
Cercopithecus aethiops (African green monkey) kidney, ATCC CRL 1586). Both cell lines were grown as monolayers in a humidified atmosphere containing 5% CO
2, the C6/36 cells at 28 °C and Vero E6 cells at 37 °C as described by Ahasan et al. [
12].
2.8. Virus Isolation in Cultured Cells
Virus isolation attempts were made only from the spleens of animals OV610, OV617, OV862, and OV867 as they tested positive for EHDV-2 in primary RT-PCR screens (
Table 1) and we thus assumed that virus was the causative agent of these animals’ deaths, and as mentioned above, EHDV can be isolated from spleen of white-tailed deer that have succumbed to the virus [
8,
11]. Similarly, EHDV was also considered the etiologic agent of OV926′s death based on gross examination of organs upon necropsy performed in the field. As OV926 was decomposing by the time of field necropsy, only the spleen was used for virus isolation attempts. For this animal, RT-PCR screens for EHDV lacked reproducibility; the first test generated a weak EHDV-specific positive PCR amplicon, but three follow-up tests generated negative results. In contrast, virus isolation attempts were made from blood, heart, kidney, liver, lung, small intestine, and spleen tissues obtained from animal OV682, as younger deer (3 months to 1 year old) are significantly more susceptible to BTV and EHDV, and this animal was 5 years old and there was a strong possibility that the etiologic agent was a virus other than BTV or EHDV. Moreover, BTV and EHDV vgRNAs were not detected by initial RT-PCR screens of RNA purified from its spleen, raising further suspicions that a different virus was the causative agent.
For all animals, virus isolation was attempted from tissue homogenates prepared as follows: portions of thawed tissues (heart, kidney, lung, liver, spleen, or small intestine) were aseptically weighed then homogenized in sterile phosphate-buffered saline (PBS) using a sterile manual tissue grinder (Fisher Scientific, Waltham, Massachusetts, USA) to form 10% (w/v) homogenates. After clearing of debris by low-speed centrifugation (5 min at 1500× g), the supernatants were filtered through a sterile 0.45 µm pore-size polyvinylidene fluoride filter (Fisher Scientific, Cat. Number 09-720-4) to remove bacteria and other particulates, and the resulting filtrates stored at −80 °C until further use. An aliquot of thawed whole blood from animal OV682 was also diluted 1:10, but was not filtered, for virus isolation attempts.
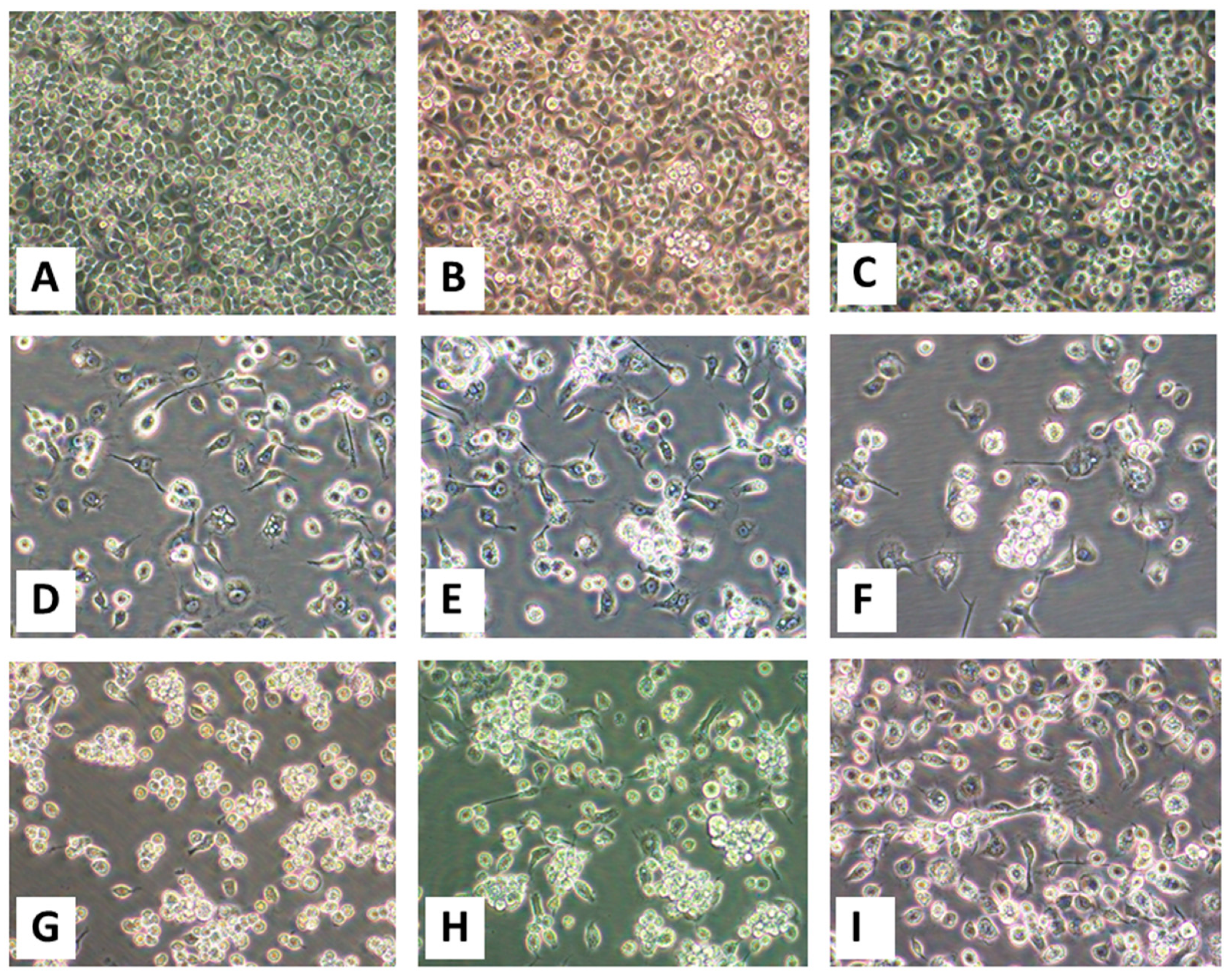
The 50-µL aliquots of filtered tissue homogenates and diluted blood from animal OV682 were inoculated onto subconfluent C6/36 and Vero E6 cells. Non-inoculated (mock-inoculated) cells were maintained in parallel as controls. The cells were re-fed every three days but monitored daily for mortality and virus-induced cytopathic effects (CPEs) for 30 days. In our work, if no CPEs are observed within the first 30 days, a second passage is routinely performed and the cells observed for another 30 days before being considered negative for virus isolation. Otherwise, after CPEs are observed in 50% of the inoculated C6/36 or Vero E6 cells, the infected cells are scraped and harvested along with the spent cell growth medium and stored at −80 °C for future analyses.
2.9. Sequencing of Virus Genomes
Virus identity and determination of the complete coding sequence (CDS) of all 10 genomes of the EHDVs and novel orbiviruses of this work were achieved through a next-generation sequencing (NGS) process as described previously [
8,
9,
10,
11,
12]. Briefly, after approximately 50% of cultured cells inoculated with tissue homogenate displayed virus-induced CPEs, vgRNAs were extracted from virions in spent medium using a QIAamp viral RNA minikit (Qiagen). A cDNA library was thereafter constructed using a NEBNext Ultra RNA library prep kit (New England Biolabs, Ipswich, Massachusetts, USA) and sequenced on an Illumina MiSeq sequencer (Illumina, Inc., San Diego, California, USA). Details regarding the EHDV sequences obtained in this study will be included in a separate report to be presented elsewhere that presents a comprehensive analysis of EHDV genomes from dead Florida farmed deer that we have analyzed over the past three years.
2.10. Genome Assembly and Annotation of Virus Sequences
For each virus, paired-end reads obtained from the Illumina MiSeq were merged and quality trimmed in CLC Genomic Workbench v10.1.1 (Qiagen). As the viruses were isolated in C6/36 cells, sequence analyses were simplified by removal of host (
Aedes albopictus) sequences deposited under GenBank accession number (GB number) MNAF00000000.2 by using Kraken v1.0 (Johns Hopkins University School of Medicine, Baltimore, Maryland, USA) [
24]. Following removal of the host sequences, de novo assembly of pair-end reads was performed in SPAdes v3.5.0 (Center for Algorithmic Biotechnology, St. Petersburg State University, St. Pertersburg, Russia) with default parameters [
25]. BLASTX (National Center for Biotechnology Information, U.S. National Library of Medicine, Bethesda, Maryland, USA) analysis of the assembled contigs were performed in CLC Genomic Workbench v10.1.1 against a custom virus genome database created from virus protein sequences retrieved from the UniProt Knowledgebase (
https://www.uniprot.org/uniprot/). Contigs representing orbivirus genome segments were validated by mapping the quality- and host-filtered reads using Bowtie2 [
26] and visually inspecting the alignments in Tablet 1.17.08.17 [
27]. The average coverage of each viral genome was estimated using Qualimap v2.2.1. Putative open reading frames (ORFs) for the orbivirus genomes were predicted using GeneMarkS (
http://exon.biology.gatech.edu/) [
28] restricting the search to virus sequences. Additionally, the functions of the genes were predicted using BLASTP searches against the NCBI non-redundant protein database.
The GB numbers of the 10 CDSs of the six novel orbiviruses and four EHDVs of this report are listed in
Table 2. In addition, for the six novel orbiviruses, information regarding the predicted gene functions of their respective segments are presented in
Supplemental Table S1 (CHeRI OrbV 1.0, 2.1, and 2.2) and
Supplemental Table S2 (CHeRI OrbV 3.1, 3.2, and 3.3).
2.11. Phylogenetic and Genetic Analyses
Maximum Likelihood (ML) phylogenetic analyses, based on the amino acid (aa) alignments of the RNA-dependent RNA polymerase (VP1) protein, the innermost sub-core capsid protein T2 (VP3), and outer capsid protein (VP2) of the CHeRI orbiviruses to those of 28 other orbiviruses whose sequences were available in GenBank (GB), were performed to assess their relationships. Amino acid sequences were aligned using (
https://mafft.cbrc.jp/alignment/software/) MAFFT [
29] and the ML analyses was performed in IQ-TREE-V 1.14.1 (
http://www.iqtree.org/) with default settings and 1000 non-parametric bootstrap analyses to test the robustness of the hypothesis [
30]. Phylogenetic trees were also constructed based on the nucleotide (nt) alignment of these genes (VP1, VP2 and VP3) following similar methods. Sequence identity matrices at both aa and nt levels were constructed using alignments of VP1, VP2, and VP3 (T2 protein) genes and proteins in Sequence Demarcation Tool v.1.2 using the MAFFT alignment option [
31]. The GB numbers of the orbivirus protein and nt sequences that were analyzed are listed in
Supplemental Table S3.
2.12. Transmission Electron Microscopy
As EHDV vgRNA was not detected by RT-PCR in a blood sample taken from animal OV682, transmission electron microscopy (TEM) of virus-infected C6/36 cells was performed to assist with virus identification before sequencing was attempted. Briefly, one nearly-confluent T75 Corning cell-culture treated flask (Thermo Fisher Scientific, Cat. number 7-202-000) of C6/36 cells was inoculated with 25 µL of first-passage cell lysate. The cells were fixed 4 days post-inoculation (dpi) in 15 mL of modified Karnovsky’s fixative (2P+2G, 2% formaldehyde prepared from paraformaldehyde and 2% glutaraldehyde in 0.1 M cacodylate buffer pH 7.4) at room temperature for 2 h, then the flask was stored at 4 °C for shipment to the University of Texas Medical Branch Electron Microscopy Laboratory (UTMB-EML). Prior to shipment on ice packs, the fixative was discarded, the cells washed once with 1 mL of ice-cold cacodylate buffer, then the wash buffer replaced with another 1 mL of ice-cold cacodylate buffer. The cells were subsequently scraped off the flask, then transferred along with the cacodylate buffer into a 15 mL conical tube, which was centrifuged at 3000× g for 10 min at 4 °C. After removing the cacodylate buffer, the pellet was resuspended in ice-cold PBS, and shipped to the UTMB-EML. At the UTMB-EML, the cell pellets were washed in cacodylate buffer and left in 2P+2G fixative overnight at 4 °C. The next day they were washed twice in cacodylate buffer, post-fixed in 1% OsO4 in 0.1 M cacodylate buffer pH 7.4, en bloc stained with 2% aqueous uranyl acetate, dehydrated in ascending concentrations of ethanol, processed through propylene oxide and embedded in Poly/Bed 812 epoxy plastic (Polysciences, Warrington, PA). Ultrathin sections (~75 nm) were cut on a Leica EM UC7 ultra-microtome (Leica Microsystems, Buffalo Grove, IL), stained with 0.4% lead citrate, and examined in a JEM-1400 electron microscope (JEOL USA, Peabody, MA) at 80 kV.
2.13. Diagnostic RT-PCR Tests Specific for CHeRI OrbV-1.0
CHeRI OrbV-1.0 was isolated in C6/36 cells from the spleen of animal OV682 but not from other tissue filtrates from the same animal. As virus isolation had initially been attempted only from spleen tissue, there was no indication whether animal OV682 was viremic for CHeRI OrbV-1.0. As a lack of CHeRI OrbV-1.0 isolation from specimens other than for spleen tissues (i.e., negative results) does not prove it was not present in the tissues that had been tested, and to gain insights on whether the virus was present in blood, two specific RT-PCR tests were devised based on the CHeRI OrbV-1.0 consensus sequences attained by NGS. Two separate RT-PCR tests were designed as a safeguard for future applications, considering gene reassortment occur among reoviruses, and mutations may occur at the RT-PCR primer-binding sites. CHeRI OrbV-1.0-specific RT-PCR tests were then used to screen aliquots of heart, kidney, liver, lung, and GI tract filtrates that had been archived at −80 °C, and frozen whole blood (
Table 1). Briefly, a sequence within CHeRI OrbV-1.0 VP1 gene was the target of the first RT-PCR test. The primers for the VP1 gene RT-PCR assay (for both cDNA synthesis and PCR) consist of CHOrbV-1.F1 (forward; 5- GGAAGAAAATCTTGACAAGGTGAG-3) and CHOrbV-1.R1 (reverse; 5-CGTATTCAAGTATTTCTCGAACC-3), which generate a 335-base pair (bp) PCR amplicon. A sequence within the CHeRI OrbV-1 VP2 gene was the target of the second RT-PCR test. The primers for the VP2 gene RT-PCR assay (for both cDNA synthesis and PCR) consist of CHOrbV-1.F2 (forward: 5-GAGTGTGATAAAGATACAGGG-3) and CHOrbV-1.R2 (reverse; 5-CTTGTCCATCCTCAATATATCCC-3), which generate a 331-bp PCR amplicon. For both sets of CHeRI OrbV-1-specific primers, the same reverse transcription and PCR were performed as: After cDNA synthesis with Omniscript reverse transcriptase (Qiagen, Inc.) at 37 °C for 60 min using both F and R primers, PCR using both F and R primers was performed using OneTAQ DNA polymerase (New England Biolabs) using the following parameters: one cycle at 94 °C for 30 s, 35 cycles of 94 °C for 15 s, 47 °C for 30 s, and 68 °C for 40 s, and one cycle 68 °C for 4 min, terminated at 4 °C (∞).
2.14. Tropism of Virus Isolates in BHK-21 Cells
Because BHK-21 cells (Mesocricetus auratus (Syrian golden hamster) kidney cells) have historically been used in attempts at arbovirus isolation in cultured cells, we tested whether the viruses of this work could be propagated in them. When they were nearly confluent, BHK-21 cells (ATCC CCL-10) that had been propagated as monolayers at 37 °C in 5% CO2 in Advanced Minimum Essential Medium (aMEM, Invitrogen) supplemented 10% FBS, GlutaMAX, PSN, and 1X nonessential amino acids (Invitrogen) and a final concentration of 1 mM sodium pyruvate (Invitrogen) were inoculated with aliquots of first-passage virus isolated in C6/36 cells. The inoculated cells were re-incubated at 37 °C in 5% CO2 and observed for 30 days with re-feeds of the cells with reduced serum (3%) aMEM every three days before being considered negative for virus isolation. Virus isolation was based on formation of virus-induced CPEs and the detection of EHDV vgRNA in spent cell culture medium by RT-PCR. In the absence of CPEs, the spent media of cells inoculated with spleen homogenate from OV682 and OV926 were tested at 5 day intervals by RT-PCR for EHDV vgRNA, and at 15 dpi by NGS.
4. Discussion
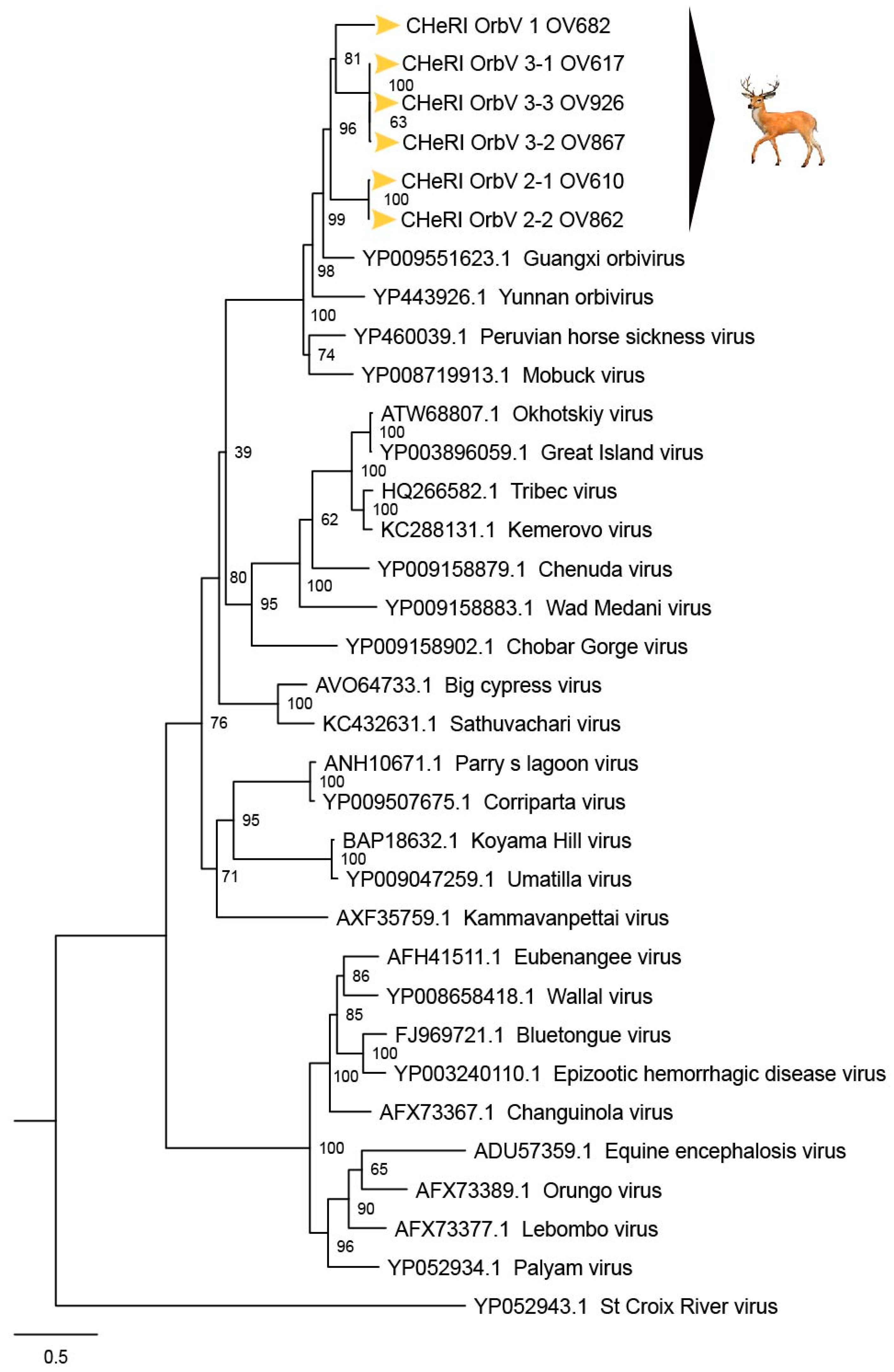
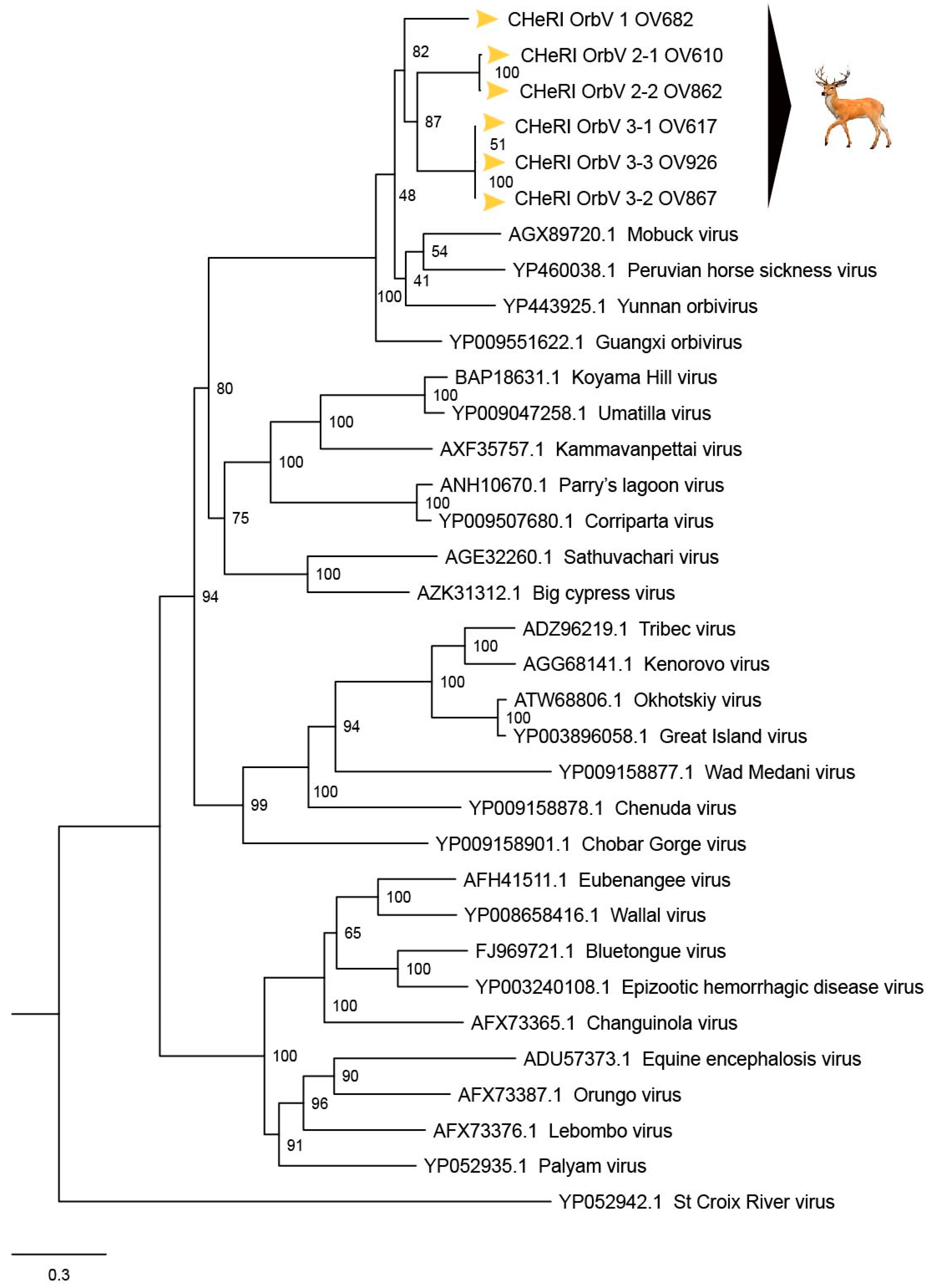
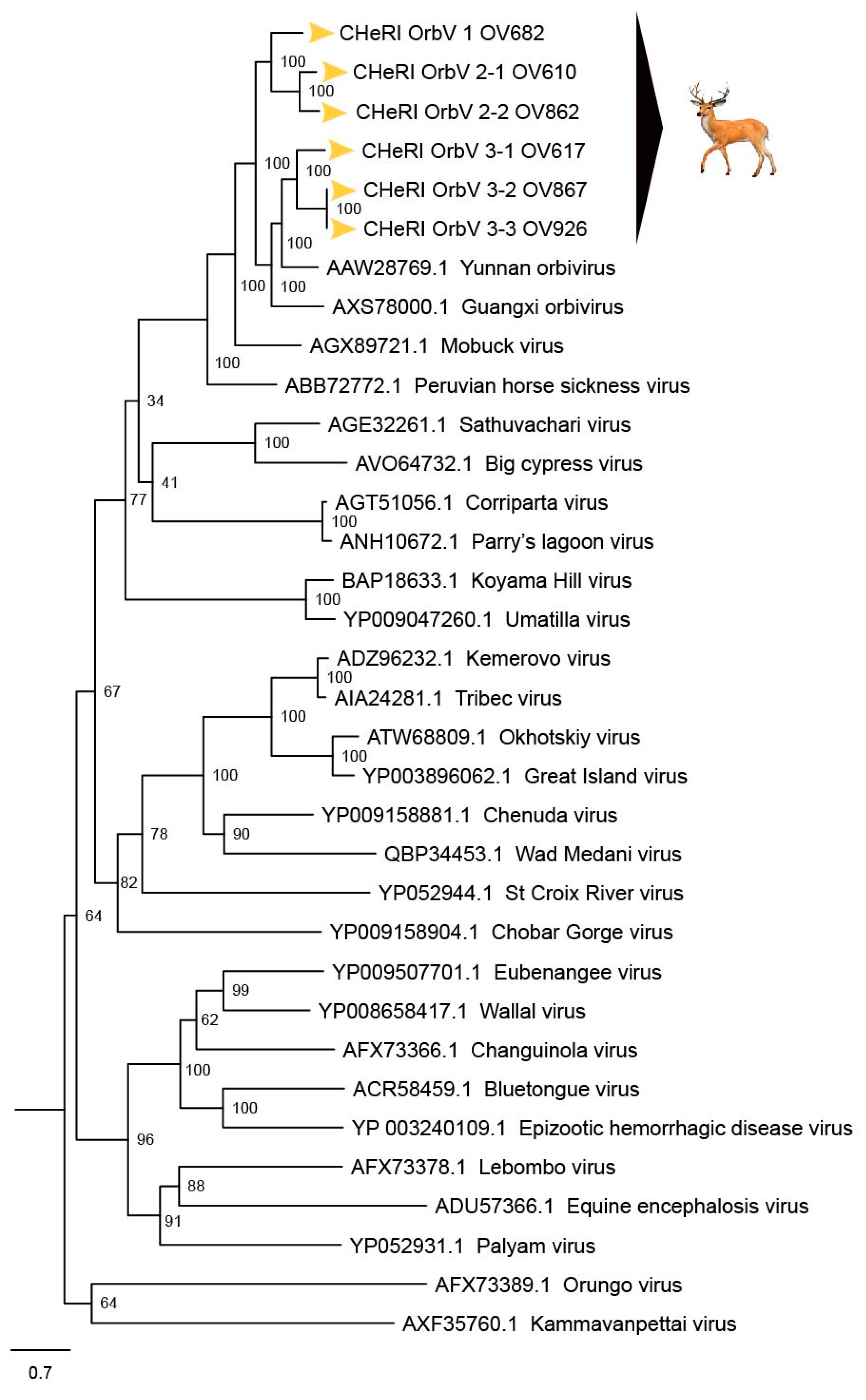
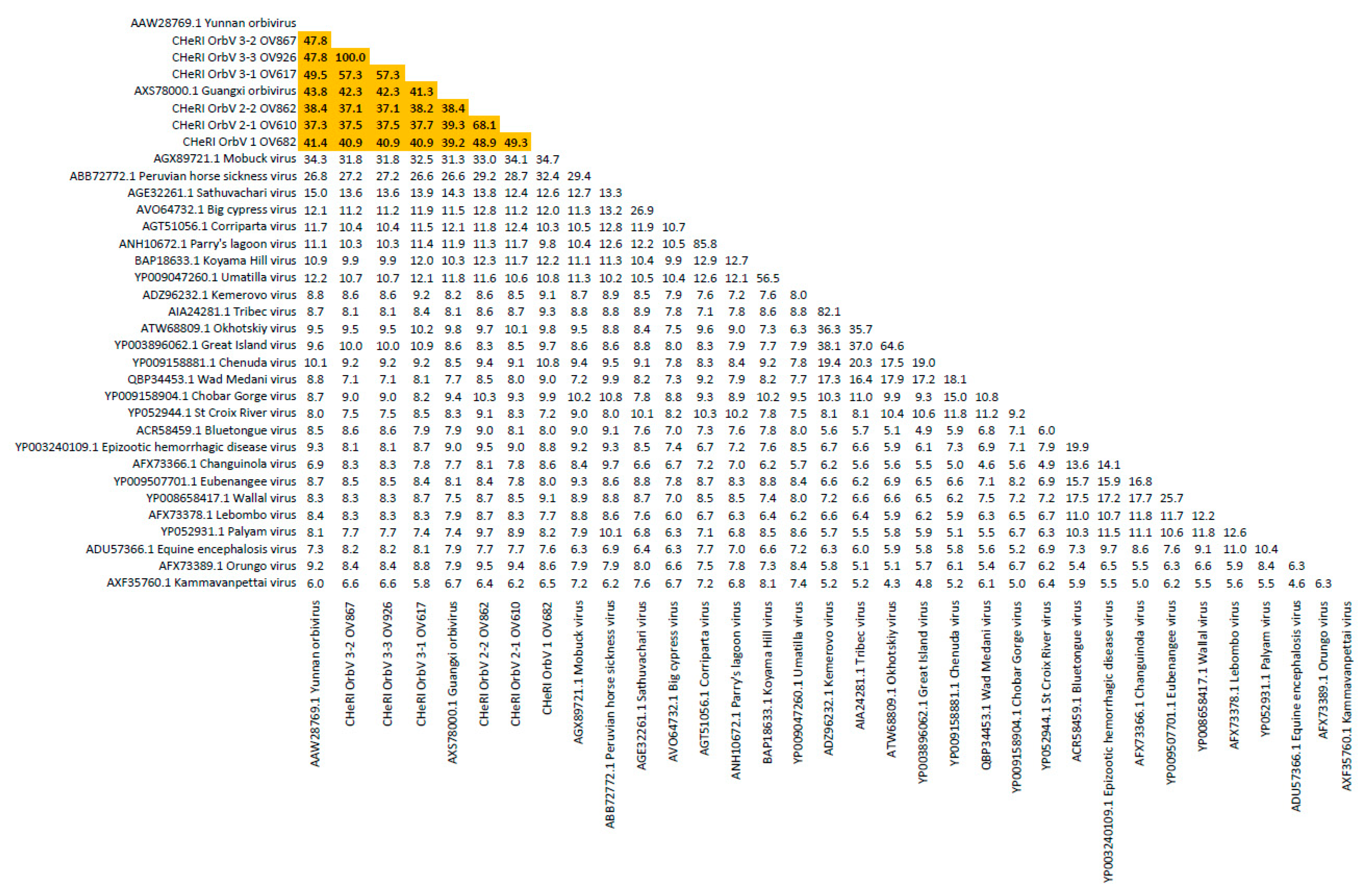
Based on ICTV criteria for orbivirus classification [
13], we reveal the discovery of three new orbivirus species along with three variants thereof in dead farmed white-tailed deer. The six novel reoviruses of this report fall within the genus
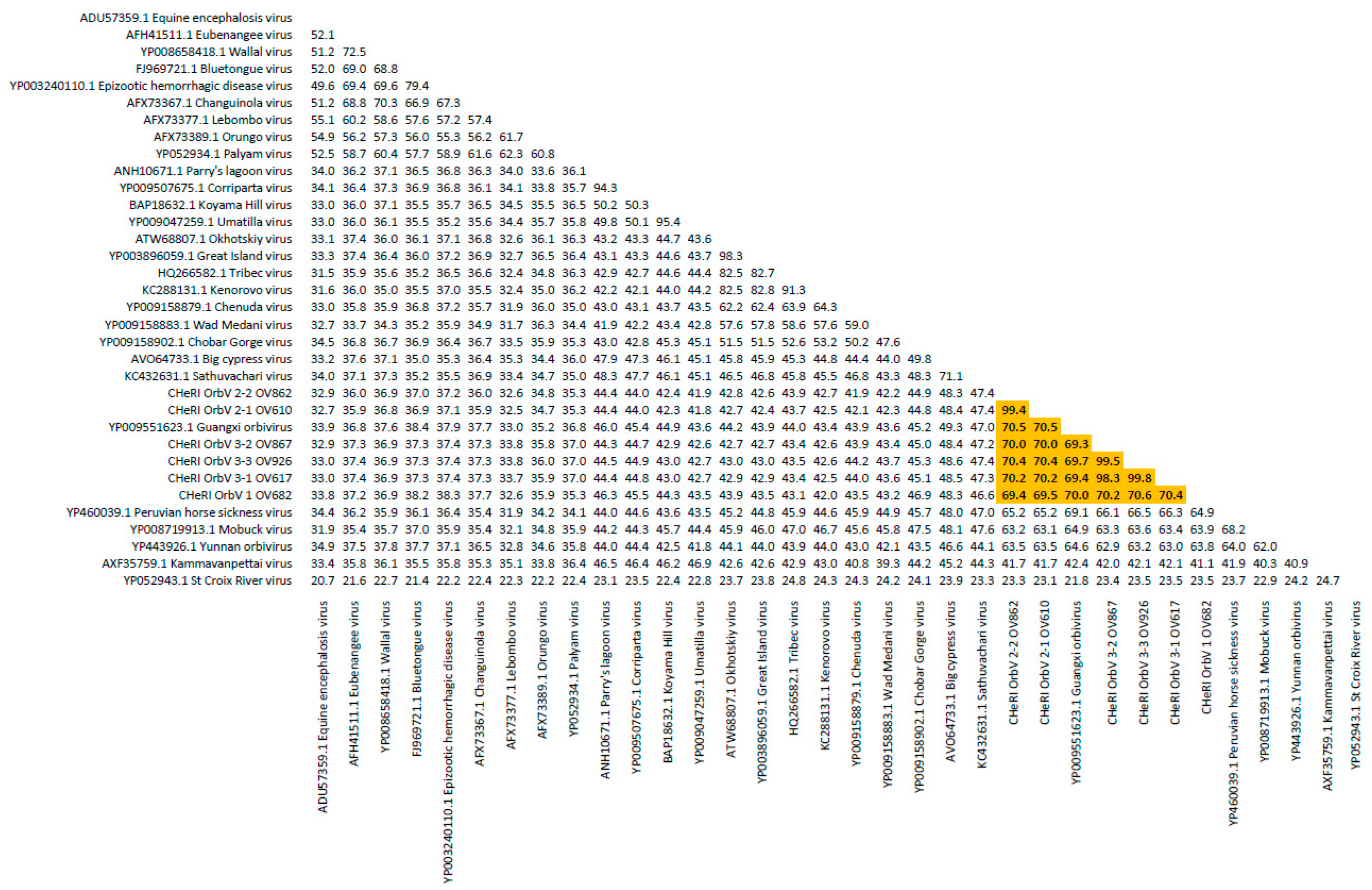
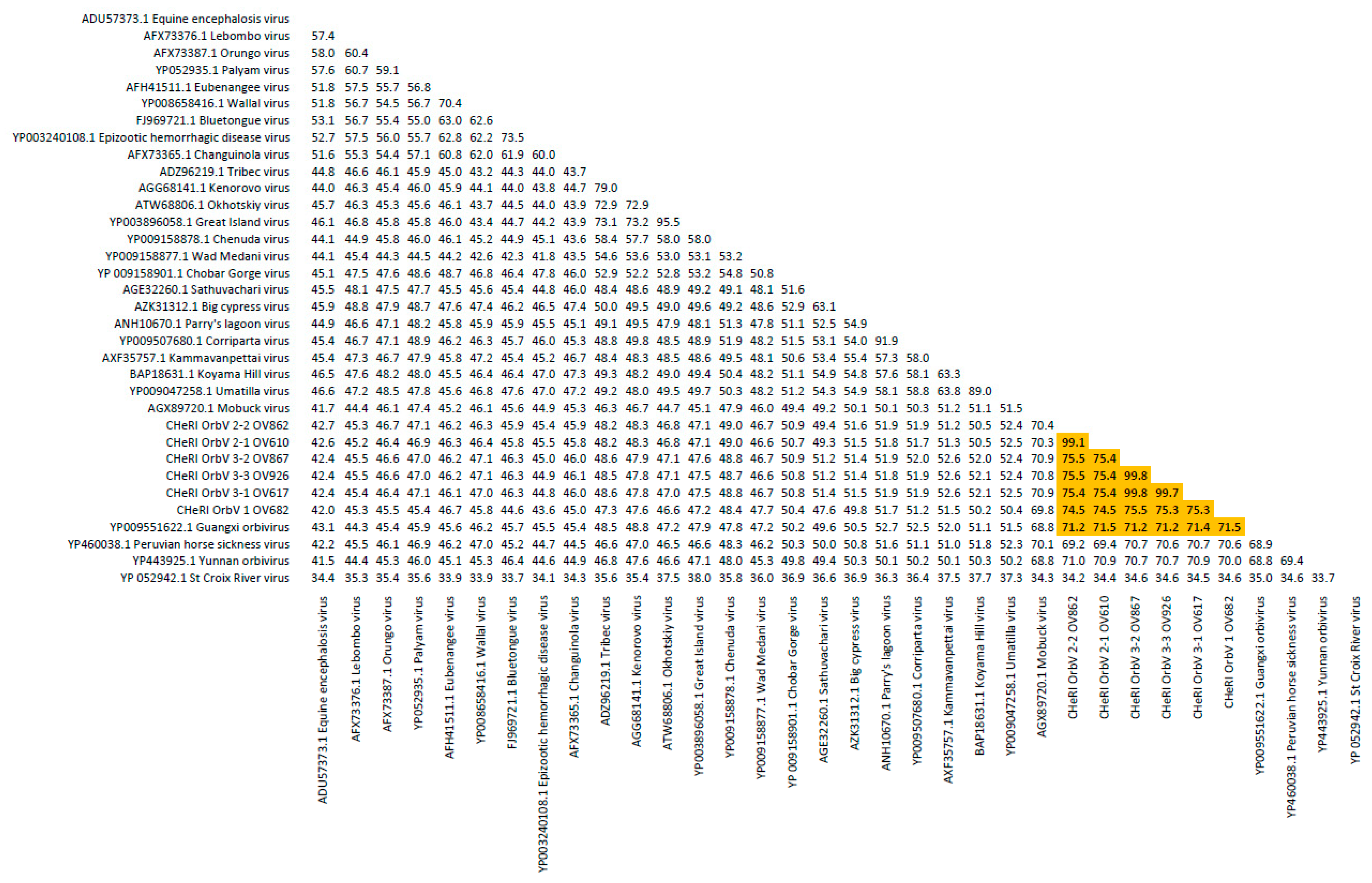
Orbivirus since their deduced RdRp (VP1) aa sequences have > 30% identities with those of other orbiviruses such as GXOV, MBV, PHSV, and YUOV (see RdRp aa sequence identity matrix,
Figure 7). Furthermore, according to ICTV criteria for orbivirus classification, T2 aa sequences are used to define orbivirus species; distinct species show < 91% sequence identities with the T2 sequences of other orbiviruses. Compared to other orbiviruses in
Figure 5, the novel orbiviruses of this report comprise three new species: CHeRI OrbV-1, CHeRI OrbV-2, and CHeRI OrbV-3. Two variants of CHeRI OrbV-2 were identified: CHeRI OrbV-2.1, and CHeRI OrbV-2.2, as were three variants of CHeRI OrbV-3: CHeRI OrbV-3.1, CHeRI OrbV-3.2, and CHeRI OrbV-3.3 (see Maximum Likelihood Phylogram of orbivirus VP2 proteins,
Figure 8). The six new orbiviruses share < 74% nt identity with other orbiviruses (
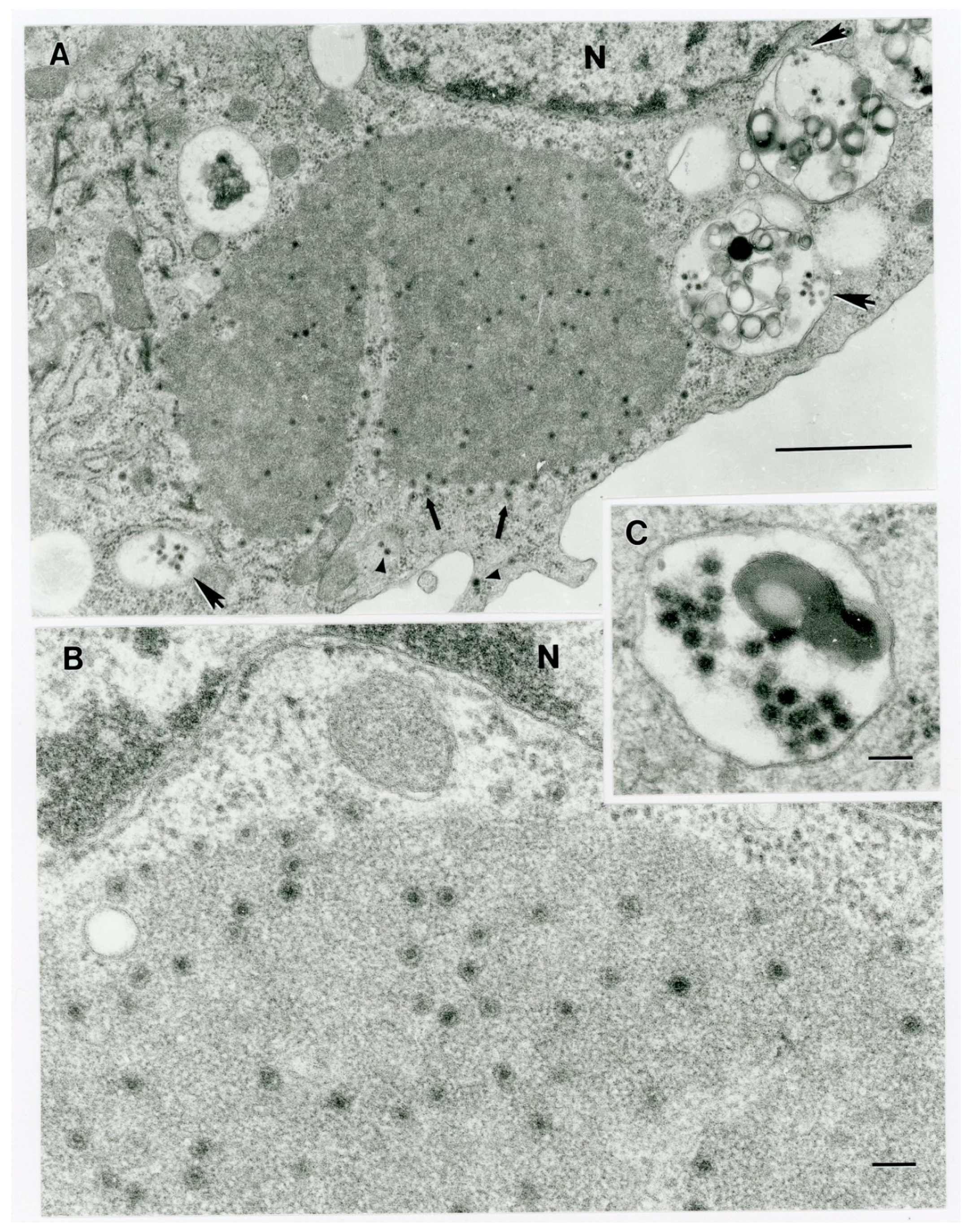
Figure 9), and that fulfils the ICTV criteria for new species. Finally, TEM studies revealed that immature CHeRI OrbV-1 particles within C6/36 cells with diameters of 50–65 nm (
Figure 3). This is consistent with observations for PHSV and YUOV, which form 55 to 60 nm immature virus particles within the cytoplasm of infected cells [
32,
33]. Taken together, the data indicate the viruses are undoubtedly orbiviruses.
Post-mortem lesions in white-tailed deer that die as a consequence of EHDV infections are characterized by hemorrhages and edema of varying severity and extent in multiple organs and tissues. Commonly affected sites include the heart and serosal surfaces of the pleural and peritoneal cavities; pulmonary edema and pericardial effusion is seen in some cases. Features suggestive of EHDV involvement were observed during gross examination of the deer of this report; in particular, lung involvement was common for all the dead deer (
Table 1). However, it was specifically noted that the animals OV682 and OV926, from which were isolated CHeRI OrbV-1 and OrbV-3.3, respectively, but no co-infecting EHDV-2 or BTV (
Table 1 and
Table 3), had normal-appearing hearts. Future studies will have to address whether those viruses do not affect cardiac tissues.
No EEEV and WNV vgRNAs were detected in specimens from any of the six deer. Moreover, neither EEEV nor WNV were isolated in cell cultures inoculated with any of the specimens tested. While negative findings do not rule out the involvement of EEEV or WNV, the preponderance of the available evidence is consistent with an orbivirus infection leading to disease and death in the six animals. Phylogenetic evaluation of the viruses of this work (
Figure 4,
Figure 6 and
Figure 8) consistently groups them with GXOV, MBV, PHSV, and YUOV. Mobuck virus was originally isolated from a white-tailed deer in Missouri, USA [
34] and later from a white-tailed deer in Florida, North America [
9]. The detection of viruses with high genetic relatedness to MBV in the dead white-tailed deer of this work appears consistent with the known biology of MBV. Guangxi virus, PHSV, and YUOV have been associated with disease in equids and cattle, and this raises the question whether the novel orbiviruses of this work are pathogens to those animals also. Another topic of interest resulting from this work is the question whether the new orbiviruses are a spill-over agents from cattle, equids, or some yet unspecified animal in Florida.
Due to the close genetic relatedness of the three new orbivirus species to the aforementioned mosquito-borne viruses, it is possible that the new viruses are also mosquito-borne viruses. Orbiviruses exemplified by YUOV usually can be isolated in mosquito (or other arthropod cells) but not in mammalian cells [
32,
33,
34,
35]. The six novel orbiviruses of this work were also not isolatable in Vero E6 or BHK-1 cells. These findings highlight the need to test other mammalian cell lines, especially bovine, equine and cervid cell lines and primary cells, for their utility as indicator cell lines for diagnostic work, and for basic research purposes. If these novel orbiviruses are found to contribute to disease, diagnostic labs need to incorporate them in their assessment of samples from moribund dear. Future development of diagnostic assays is needed to determine the prevalence of these orbiviruses in archived samples involving dear epizootics.
The prevention and control of arthropod-vectored orbiviruses is dependent upon knowledge of virus–vector–host interactions, pathogenesis, diagnostics, epidemiology, and control strategies. A better understanding of the basic virology of our new orbivirus is needed to: (1) help model current and future disease outbreaks, (2) develop vaccines, (3) develop reliable diagnostic tests, (4) develop effective vector control strategies, and (5) develop criteria and methods for domestic (enzootic) versus exotic (non-enzootic or incursive) serogrouping and topotyping. For our new viruses, transmission mechanisms in and between vertebrate hosts and arthropod vectors should be evaluated to determine their potential to emerge and threaten animal health, livestock trade, and even human health.
It is interesting that four of the six deer of this report had mixed virus infections. In each case, one of the novel orbiviruses was found together with EHDV-2. Each EHDV-2 strain was genetically distinct (the nucleotide sequences of their gene segments were not identical), indicating that the presence of EHDV-2 was not due to some sort of laboratory contaminant. Furthermore, we and others found MBV also in association with EHDV-2 [
9,
33]. An association of EHDV-2 with another orbivirus in infected tissue suggests some yet unknown interaction that may exacerbate pathology and warrants further investigation. According to our phylogenetic analyses, the novel orbiviruses group with mosquito-borne viruses whereas EHDV-2 is transmitted by
Culicoides species. This raises the question whether our understanding of the vector biology of EHDV-2 is inaccurate (i.e., that the virus can also be vectored by mosquitoes), and vice-versa, whether the novel orbiviruses of this work are vectored not by mosquitoes but by
Culicoides species. Additional research is needed to address these questions.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}