Multi-Approach Investigation Regarding the West Nile Virus Situation in Hungary, 2018

 , , ,
, , ,  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Nucleic Acid Preparation
2.3. PCR Amplification and Sequencing of Animal Samples
2.4. Molecular Diagnostics of the Human WNV Infections
2.5. Serological Diagnosis of Human WNV Cases
2.6. In Vitro Virus Propagation
2.7. Nucleotide Sequence Analysis
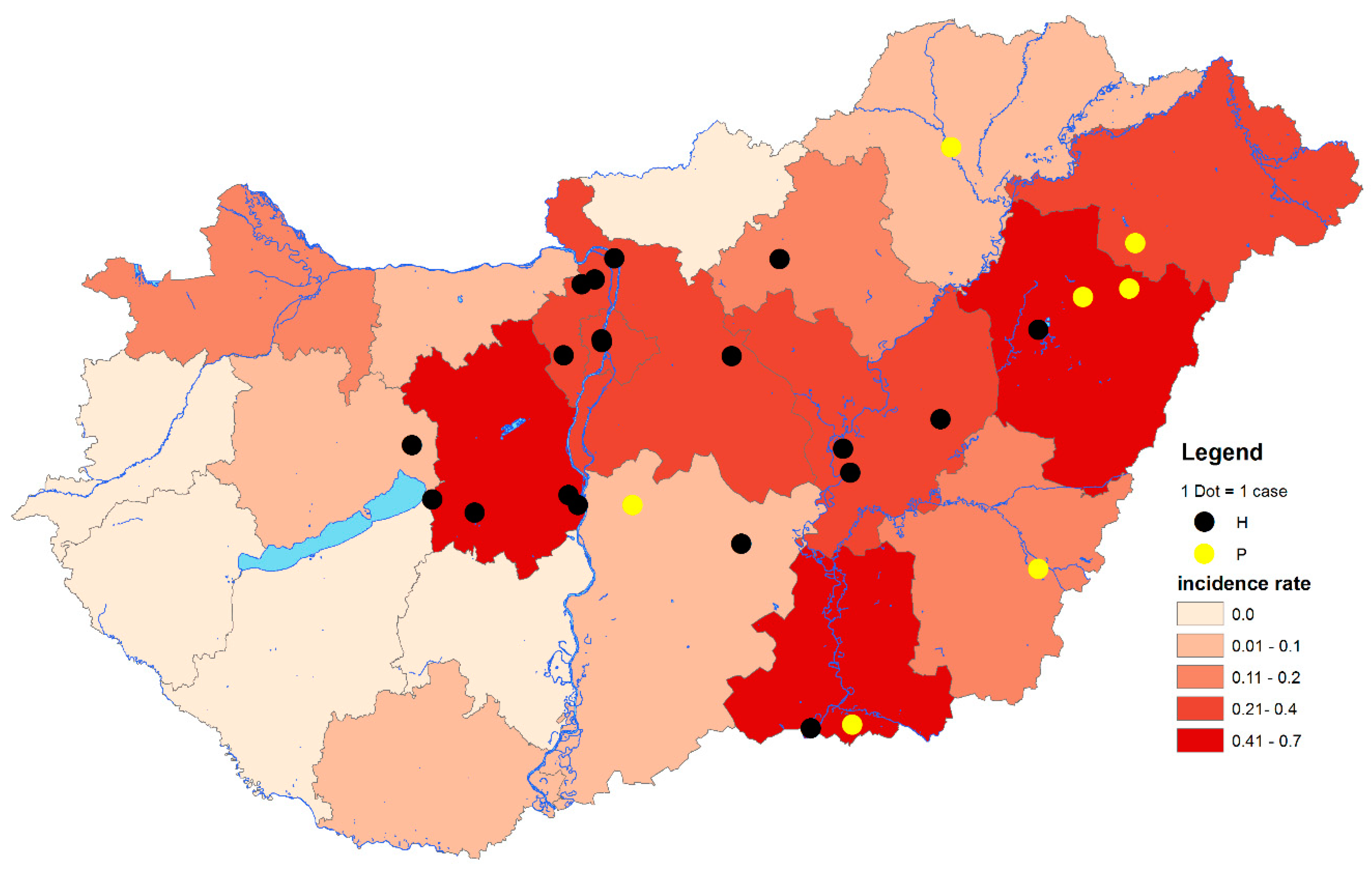
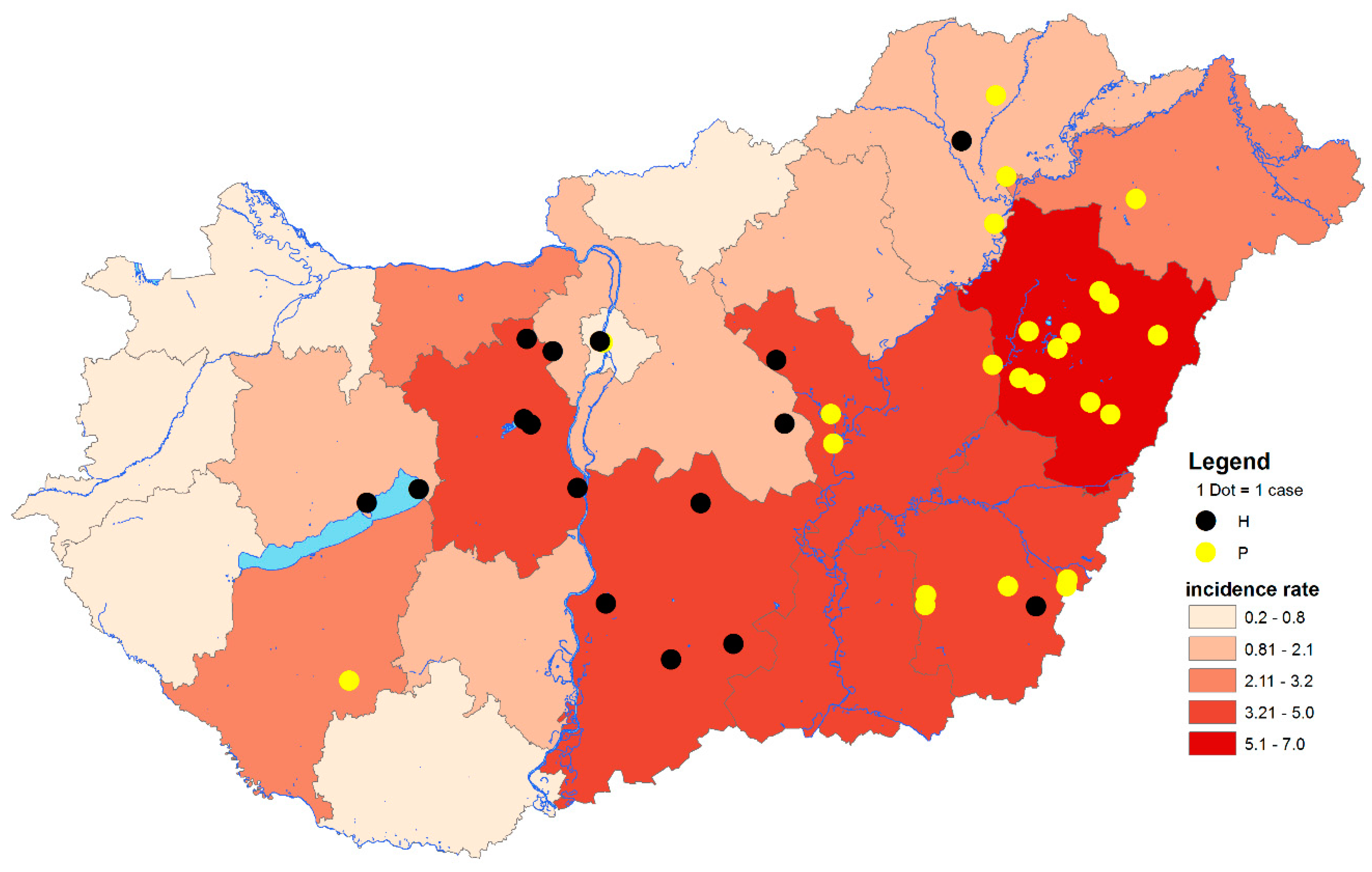
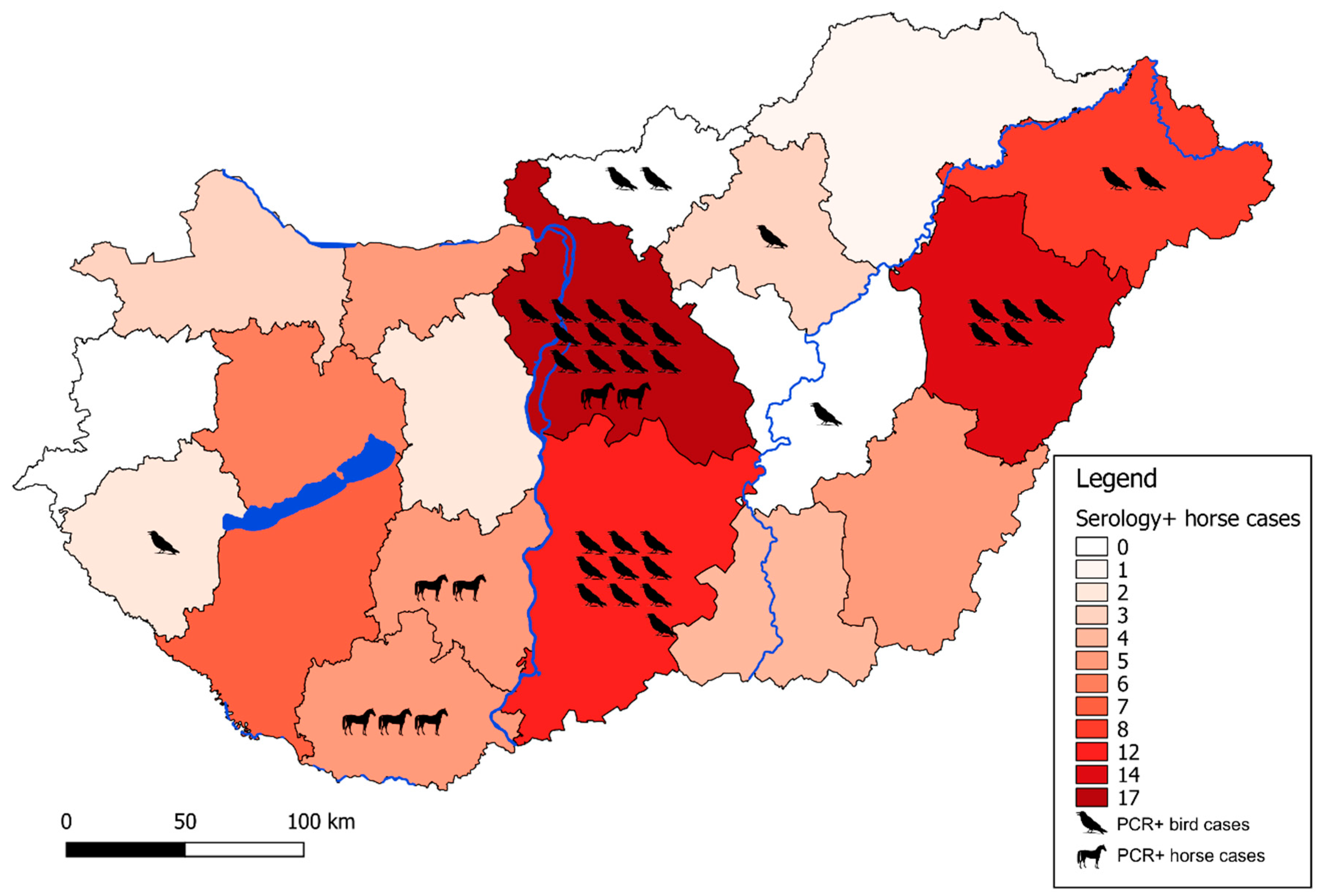
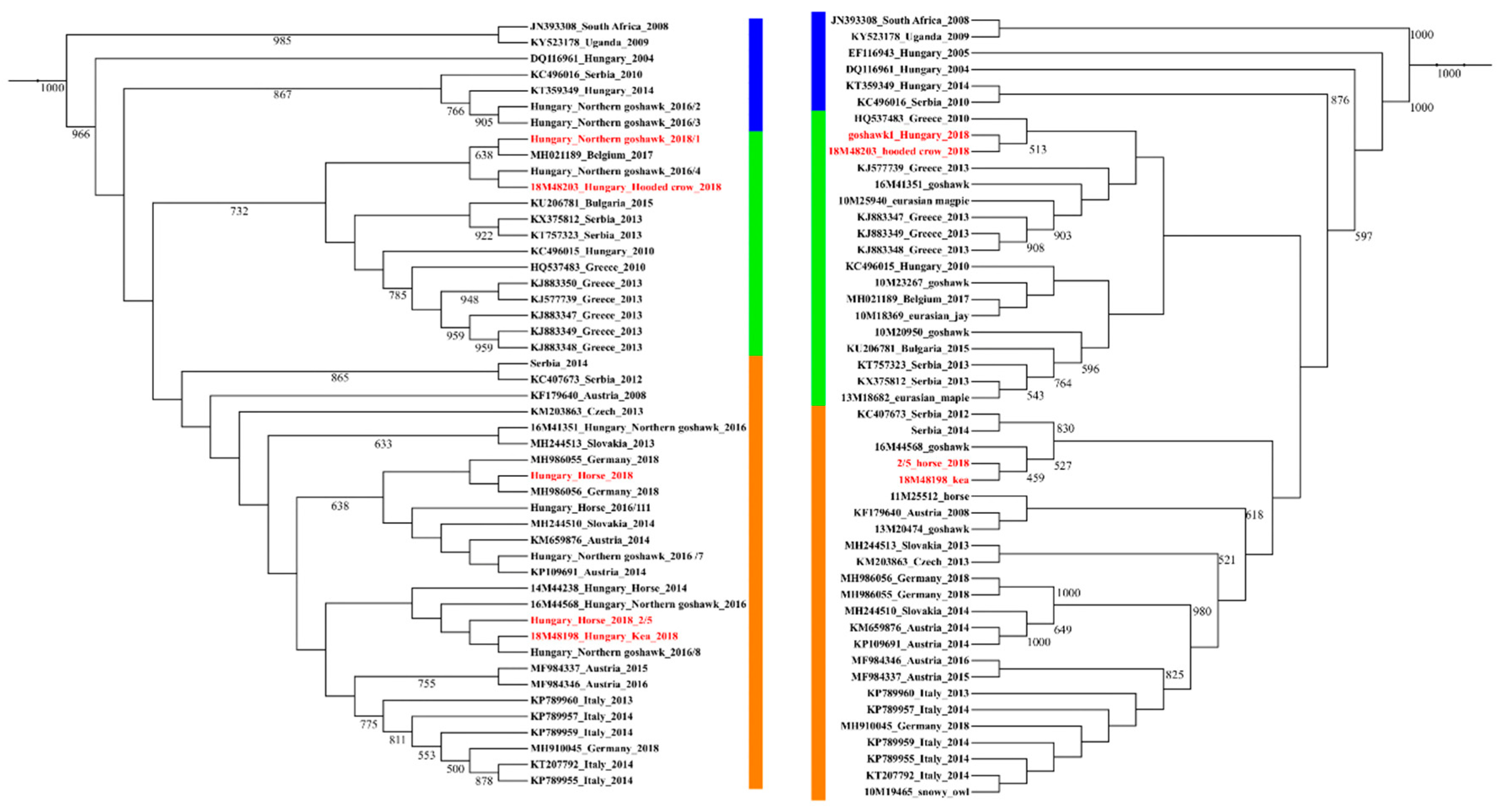
3. Results and Discussion
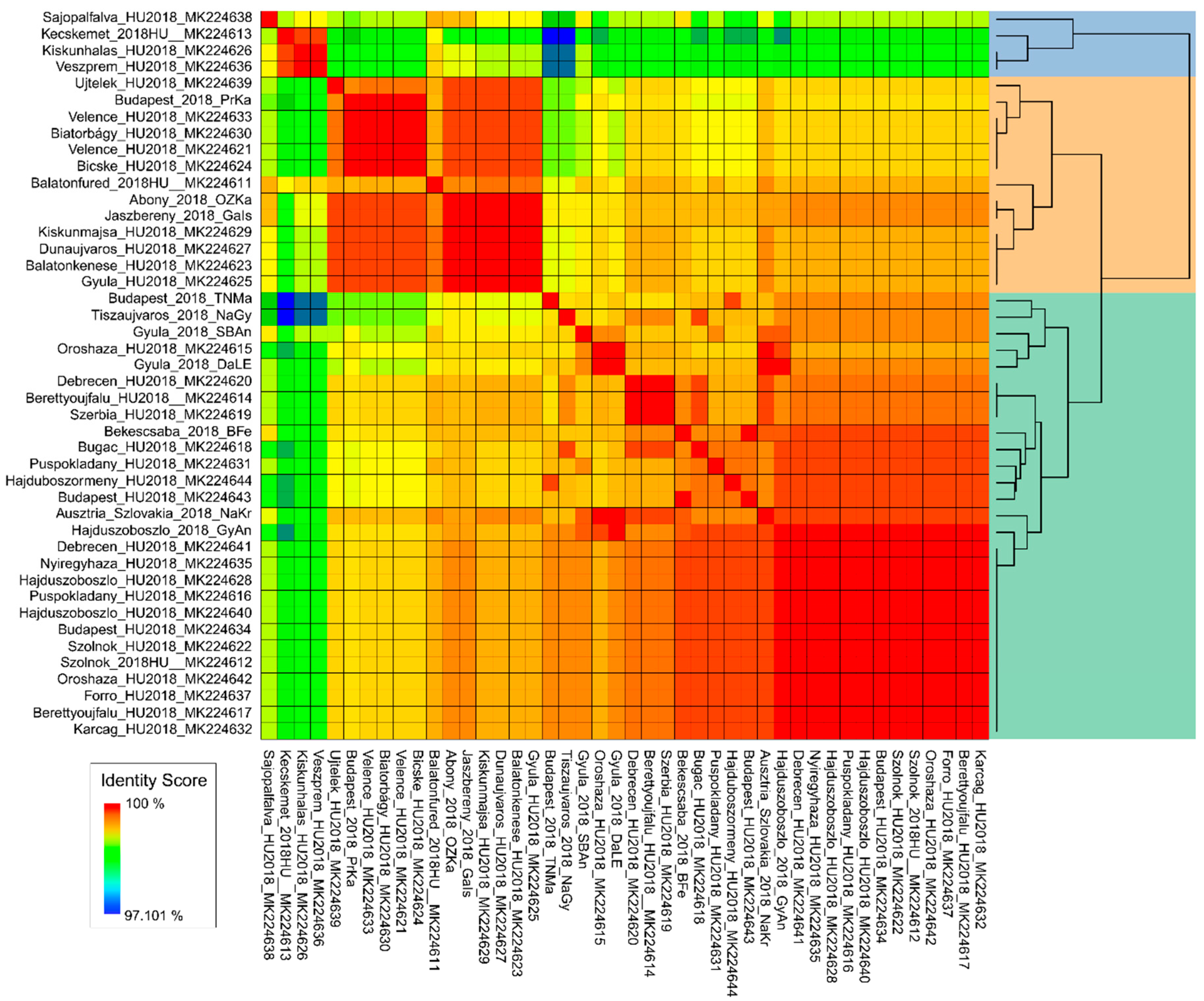
3.1. Virus Detection
3.1.1. PCR Screening
3.1.2. Equine Serologic Survey
3.1.3. In Vitro Virus Propagation
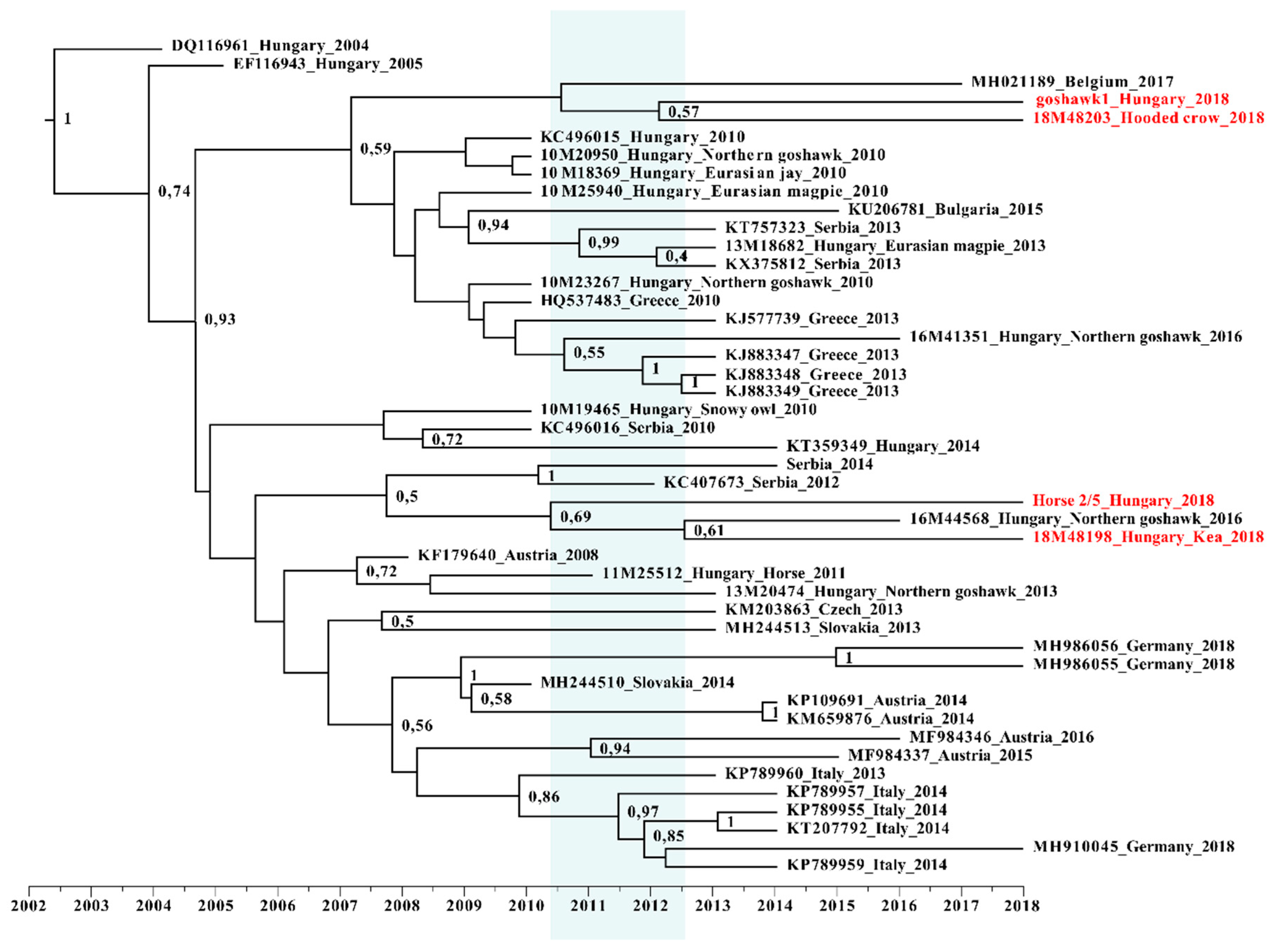
3.2. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gray, T.J.; Webb, C.E. A review of the epidemiological and clinical aspects of West Nile virus. Int. J. Gen. Med. 2014, 7, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Kemenesi, G.; Dallos, B.; Oldal, M.; Kutas, A.; Földes, F.; Németh, V.; Reiter, P.; Bakonyi, T.; Bányai, K.; Jakab, F. Putative novel lineage of West Nile virus in Uranotaenia unguiculata mosquito, Hungary. Virusdisease 2014, 25, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Bakonyi, T.; Xanthopoulou, K.; Vázquez, A.; Tenorio, A.; Nowotny, N. Genetic characterization of West Nile virus lineage 2, Greece, 2010. Emerg. Infect. Dis. 2011, 17, 920–922. [Google Scholar] [CrossRef] [PubMed]
- Venter, M.; Steyl, J.; Human, S.; Weyer, J.; Zaayman, D.; Blumberg, L.; Leman, P.A.; Paweska, J.; Swanepoel, R. Transmission of West Nile virus during horse autopsy. Emerg. Infect. Dis. 2010, 16, 573–575. [Google Scholar] [CrossRef]
- Napp, S.; Petrić, D.; Busquets, N. West Nile virus and other mosquito-borne viruses present in Eastern Europe. Pathog. Glob. Health 2018, 112, 233–248. [Google Scholar] [CrossRef]
- Ciota, A.T.; Kramer, L.D. Vector-virus interactions and transmission dynamics of West Nile virus. Viruses 2013, 5, 3021–3047. [Google Scholar] [CrossRef]
- Paphitou, N.I.; Tourvas, A.; Floridou, D.; Richter, J.; Tryfonos, C.; Christodoulou, C. The first human case of neuroinvasive West Nile virus infection identified in Cyprus. J. Infect. Public Health 2017, 10, 891–893. [Google Scholar] [CrossRef]
- Zannoli, S.; Sambri, V. West Nile Virus and Usutu Virus Co-Circulation in Europe: Epidemiology and Implications. Microorganisms 2019, 7, 184. [Google Scholar] [CrossRef]
- Ulbert, S. West Nile virus: The complex biology of an emerging pathogen. Intervirology 2011, 54, 171–184. [Google Scholar] [CrossRef]
- Bakonyi, T.; Ivanics, E.; Erdélyi, K.; Ursu, K.; Ferenczi, E.; Weissenböck, H.; Nowotny, N. Lineage 1 and 2 strains of encephalitic West Nile virus, central Europe. Emerg. Infect. Dis. 2006, 12, 618–623. [Google Scholar] [CrossRef]
- Bakonyi, T.; Ferenczi, E.; Erdélyi, K.; Kutasi, O.; Csörgő, T.; Seidel, B.; Weissenböck, H.; Brugger, K.; Bán, E.; Nowotny, N. Explosive spread of a neuroinvasive lineage 2 West Nile virus in Central Europe, 2008/2009. Vet. Microbiol. 2013, 165, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Triana, L.M.; Jeffries, C.L.; Mansfield, K.L.; Carnell, G.; Fooks, A.R.; Johnson, N. Emergence of west nile virus lineage 2 in europe: A review on the introduction and spread of a mosquito-borne disease. Front. Public Health 2014, 2, 271. [Google Scholar] [CrossRef]
- Kurolt, I.C.; Krajinović, V.; Topić, A.; Kuzman, I.; Baršić, B.; Markotić, A. First molecular analysis of West Nile virus during the 2013 outbreak in Croatia. Virus Res. 2014, 189, 63–66. [Google Scholar] [CrossRef] [PubMed]
- ECDC: European Centre for Disease Prevention and Control. Available online: https://www.ecdc.europa.eu/en/news-events/epidemiological-update-west-nile-virus-transmission-season-europe-2017 (accessed on 13 November 2019).
- ECDC: European Centre for Disease Prevention and Control. Available online: https://www.ecdc.europa.eu/en/news-events/epidemiological-update-west-nile-virus-transmission-season-europe-2018 (accessed on 13 November 2019).
- Kemenesi, G.; Buzás, D.; Zana, B.; Kurucz, K.; Krtinic, B.; Kepner, A.; Földes, F.; Jakab, F. First genetic characterization of Usutu virus from Culex pipiens mosquitoes Serbia, 2014. Infect. Genet. Evol. 2018, 63, 58–61. [Google Scholar] [CrossRef]
- Becker, N.; Zgomba, M.; Petric, D.; Dahl, C.; Boase, C.; Lane, J.; Kaiser, A. Mosquitoes and Their Control, 1st ed.; Kluwer Academic/Plenum Publisher: New York, NY, USA, 2003; Volume 498. [Google Scholar]
- Weidinger, P.; Kolodziejek, J.; Bakonyi, T.; Brunthaler, R.; Erdélyi, K.; Weissenböck, H.; Nowotny, N. Different dynamics of Usutu virus infections in Austria and Hungary, 2017–2018. Transbound. Emerg. Dis. 2019, 67, 298–307. [Google Scholar] [CrossRef]
- Tang, Y.; Anne Hapip, C.; Liu, B.; Fang, C.T. Highly sensitive TaqMan RT-PCR assay for detection and quantification of both lineages of West Nile virus RNA. J. Clin. Virol. 2006, 36, 177–182. [Google Scholar] [CrossRef]
- Vázquez, A.; Sánchez-Seco, M.P.; Palacios, G.; Molero, F.; Reyes, N.; Ruiz, S.; Aranda, C.; Marqués, E.; Escosa, R.; Moreno, J.; et al. Novel flaviviruses detected in different species of mosquitoes in Spain. Vector-Borne Zoonotic Dis. 2012, 12, 223–229. [Google Scholar] [CrossRef]
- Linke, S.; Ellerbrok, H.; Niedrig, M.; Nitsche, A.; Pauli, G. Detection of West Nile virus lineages 1 and 2 by real-time PCR. J. Virol. Methods 2007, 146, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Chaskopoulou, A.; Dovas, C.; Chaintoutis, S.; Bouzalas, I.; Ara, G.; Papanastassopoulou, M. Evidence of enzootic circulation of West Nile virus (Nea Santa-Greece-2010, lineage 2), Greece, May to July 2011. Eurosurveillance 2011, 16, 19933. [Google Scholar] [PubMed]
- Balogh, Z.; Egyed, L.; Ferenczi, E.; Bán, E.; Szomor, K.N.; Takács, M.; Berencsi, G. Experimental infection of goats with tick-borne encephalitis virus and the possibilities to prevent virus transmission by raw goat milk. Intervirology 2012, 55, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 13 November 2019).
- Nagy, A.; Mezei, E.; Nagy, O.; Bakonyi, T.; Csonka, N.; Kaposi, M.; Koroknai, A.; Szomor, K.; Rigó, Z.; Molnár, Z.; et al. Extraordinary increase in West Nile virus cases and first confirmed human Usutu virus infection in Hungary, 2018. Eurosurveillance 2019, 24, 1900038. [Google Scholar] [CrossRef] [PubMed]
- Erdélyi, K.; Ursu, K.; Ferenczi, E.; Szeredi, L.; Rátz, F.; Skáre, J.; Bakonyi, T. Clinical and pathologic features of lineage 2 West Nile virus infections in birds of prey in Hungary. Vector-Borne Zoonotic Dis. 2007, 7, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Hubálek, Z.; Kosina, M.; Rudolf, I.; Mendel, J.; Straková, P.; Tomešek, M. Mortality of Goshawks (Accipiter gentilis) Due to West Nile Virus Lineage 2. Vector-Borne Zoonotic Dis. 2018, 18, 624–627. [Google Scholar] [CrossRef]
- Rizzoli, A.; Bolzoni, L.; Chadwick, E.A.; Capelli, G.; Montarsi, F.; Grisenti, M.; de la Puente, J.M.; Muñoz, J.; Figuerola, J.; Soriguer, R.; et al. Understanding West Nile virus ecology in Europe: Culex pipiens host feeding preference in a hotspot of virus emergence. Parasit. Vectors 2015, 8, 213. [Google Scholar] [CrossRef]
- Napp, S.; Montalvo, T.; Piñol-Baena, C.; Gómez-Martín, M.B.; Nicolás-Francisco, O.; Soler, M.; Busquets, N. Usefulness of Eurasian Magpies (Pica pica) for West Nile virus Surveillance in Non-Endemic and Endemic Situations. Viruses 2019, 11, 716. [Google Scholar] [CrossRef]
- Rudolf, I.; Betášová, L.; Blažejová, H.; Venclíková, K.; Straková, P.; Šebesta, O.; Mendel, J.; Bakonyi, T.; Schaffner, F.; Nowotny, N.; et al. West Nile virus in overwintering mosquitoes, central Europe. Parasit. Vectors 2017, 10, 452. [Google Scholar] [CrossRef]
- Bakonyi, T.; Gajdon, G.K.; Schwing, R.; Vogl, W.; Häbich, A.C.; Thaller, D.; Weissenböck, H.; Rudolf, I.; Hubálek, Z.; Nowotny, N. Chronic West Nile virus infection in kea (Nestor notabilis). Vet. Microbiol. 2016, 183, 135–139. [Google Scholar] [CrossRef]
- Chaintoutis, S.C.; Papa, A.; Pervanidou, D.; Dovas, C.I. Evolutionary dynamics of lineage 2 West Nile virus in Europe, 2004–2018: Phylogeny, selection pressure and phylogeography. Mol. Phylogenet. Evol. 2019, 141, 106617. [Google Scholar] [CrossRef] [PubMed]
- Wollants, E.; Smolders, D.; Naesens, R.; Bruynseels, P.; Lagrou, K.; Matthijnssens, J.; Van Ranst, M. Use of Next-Generation Sequencing for Diagnosis of West Nile Virus Infection in Patient Returning to Belgium from Hungary. Emerg. Infect. Dis. 2018, 24, 2380–2382. [Google Scholar] [CrossRef] [PubMed]
- Veo, C.; Della Ventura, C.; Moreno, A.; Rovida, F.; Percivalle, E.; Canziani, S.; Torri, D.; Calzolari, M.; Baldanti, F.; Galli, M.; et al. Evolutionary Dynamics of the Lineage 2 West Nile Virus That Caused the Largest European Epidemic: Italy 2011–2018. Viruses 2019, 11, 814. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zana, B.; Erdélyi, K.; Nagy, A.; Mezei, E.; Nagy, O.; Takács, M.; Bakonyi, T.; Forgách, P.; Korbacska-Kutasi, O.; Fehér, O.; et al. Multi-Approach Investigation Regarding the West Nile Virus Situation in Hungary, 2018. Viruses 2020, 12, 123. https://doi.org/10.3390/v12010123
Zana B, Erdélyi K, Nagy A, Mezei E, Nagy O, Takács M, Bakonyi T, Forgách P, Korbacska-Kutasi O, Fehér O, et al. Multi-Approach Investigation Regarding the West Nile Virus Situation in Hungary, 2018. Viruses. 2020; 12(1):123. https://doi.org/10.3390/v12010123
Chicago/Turabian StyleZana, Brigitta, Károly Erdélyi, Anna Nagy, Eszter Mezei, Orsolya Nagy, Mária Takács, Tamás Bakonyi, Petra Forgách, Orsolya Korbacska-Kutasi, Orsolya Fehér, and et al. 2020. "Multi-Approach Investigation Regarding the West Nile Virus Situation in Hungary, 2018" Viruses 12, no. 1: 123. https://doi.org/10.3390/v12010123
APA StyleZana, B., Erdélyi, K., Nagy, A., Mezei, E., Nagy, O., Takács, M., Bakonyi, T., Forgách, P., Korbacska-Kutasi, O., Fehér, O., Malik, P., Ursu, K., Kertész, P., Kepner, A., Martina, M., Süli, T., Lanszki, Z., Tóth, G. E., Kuczmog, A., ... Kemenesi, G. (2020). Multi-Approach Investigation Regarding the West Nile Virus Situation in Hungary, 2018. Viruses, 12(1), 123. https://doi.org/10.3390/v12010123