Modeling Challenges of Ebola Virus–Host Dynamics during Infection and Treatment

 ,
, 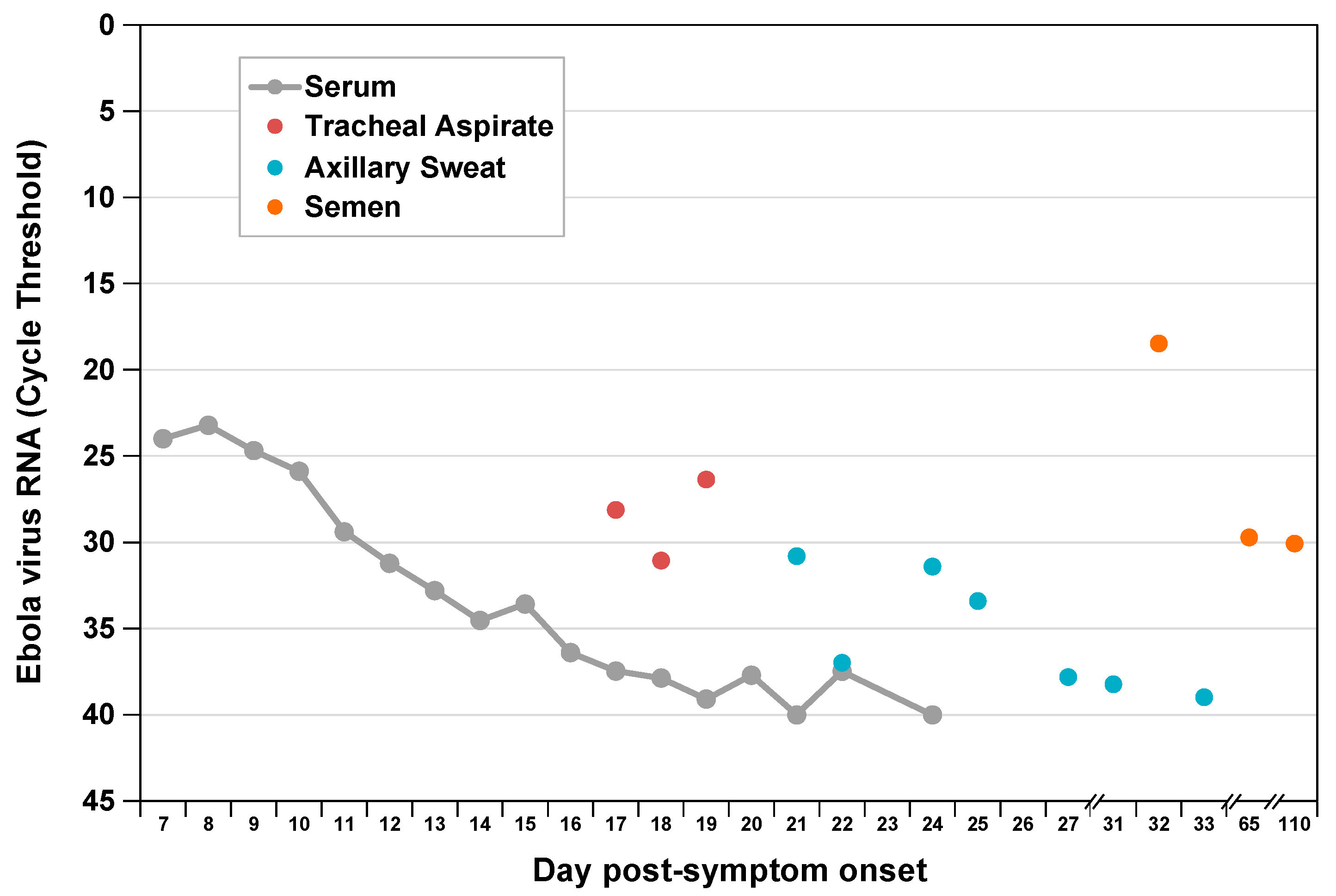{kind=link}
{kind=link}
{kind=link}
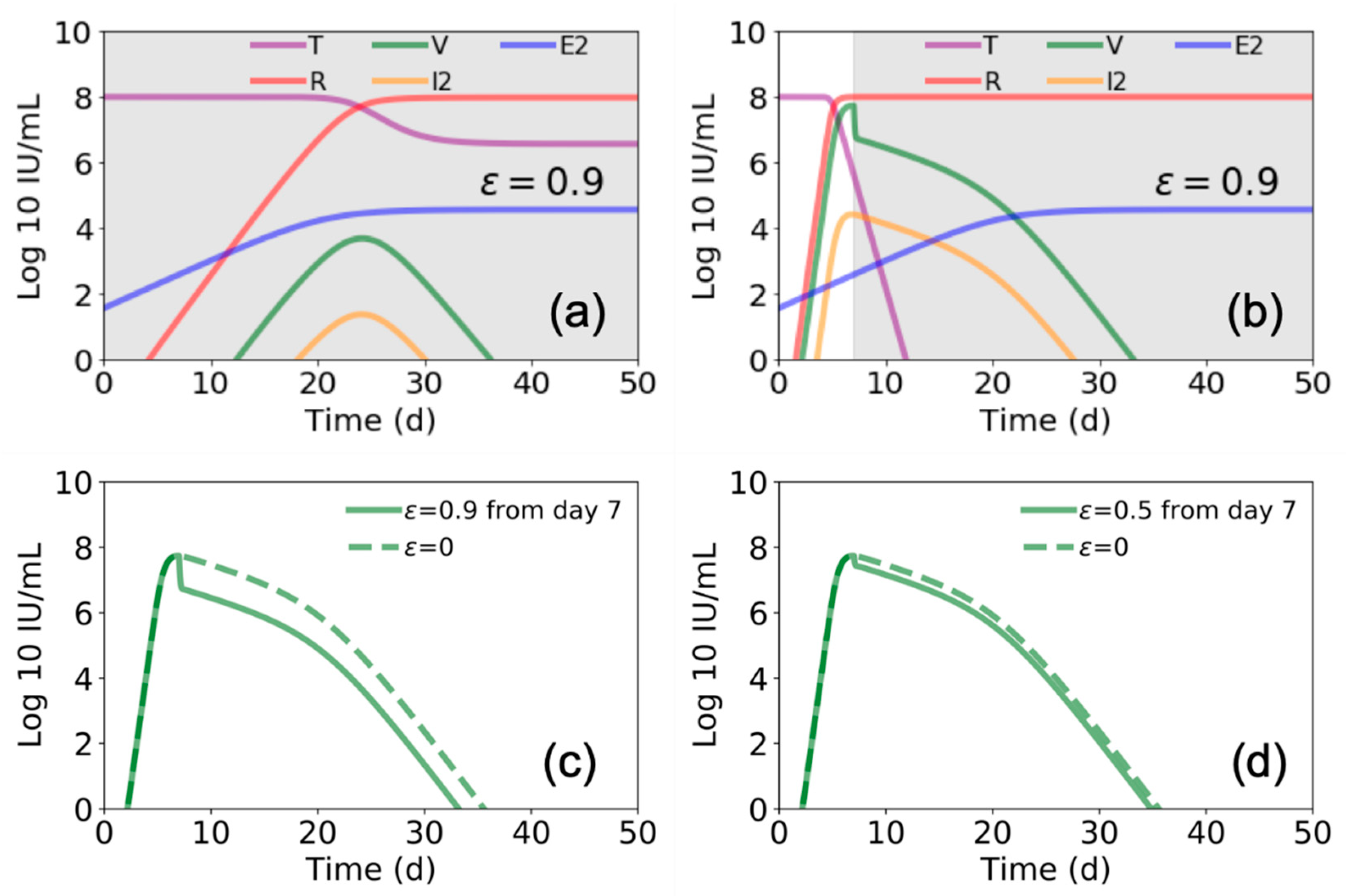{kind=link}
Abstract
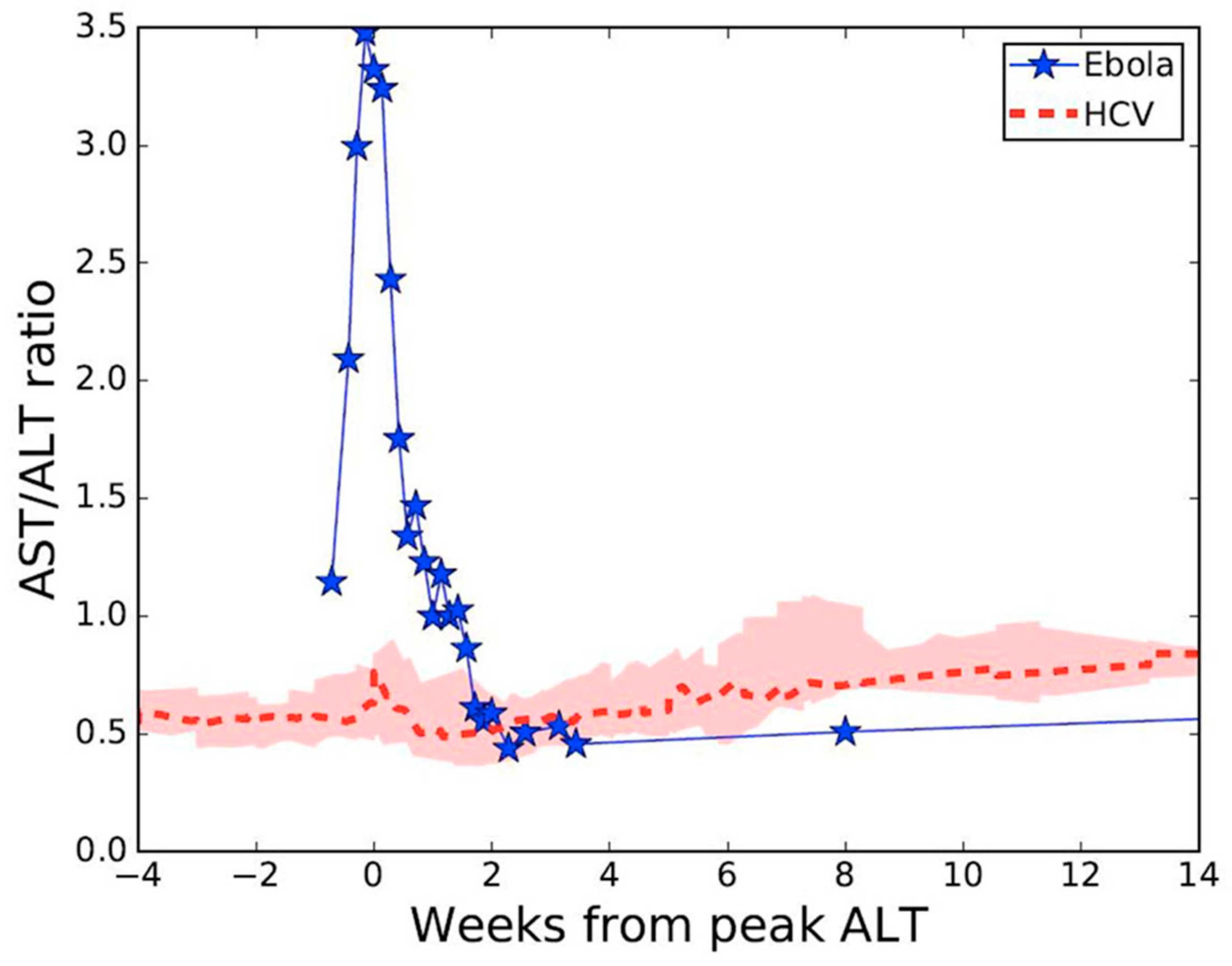
1. Introduction
2. Material and Methods
2.1. Patients
2.2. Mathematical Modeling
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Erb-Alvarez, J.; Wendelboe, A.M.; Chertow, D.S. Ebola Virus in the Democratic Republic of the Congo: Advances and Remaining Obstacles in Epidemic Control, Clinical Care, and Biomedical Research. Chest 2019. [Google Scholar] [CrossRef]
- Nguyen, V.K.; Hernandez-Vargas, E.A. Windows of opportunity for Ebola virus infection treatment and vaccination. Sci. Rep. 2017, 7, 8975. [Google Scholar] [CrossRef] [PubMed]
- Martyushev, A.; Nakaoka, S.; Sato, K.; Noda, T.; Iwami, S. Modelling Ebola virus dynamics: Implications for therapy. Antivir. Res. 2016, 135, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Madelain, V.; Baize, S.; Jacquot, F.; Reynard, S.; Fizet, A.; Barron, S.; Solas, C.; Lacarelle, B.; Carbonnelle, C.; Mentre, F.; et al. Ebola viral dynamics in nonhuman primates provides insights into virus immuno-pathogenesis and antiviral strategies. Nat. Commun. 2018, 9, 4013. [Google Scholar] [CrossRef] [PubMed]
- Chertow, D.S.; Nath, A.; Suffredini, A.F.; Danner, R.L.; Reich, D.S.; Bishop, R.J.; Childs, R.W.; Arai, A.E.; Palmore, T.N.; Lane, H.C.; et al. Severe Meningoencephalitis in a Case of Ebola Virus Disease: A Case Report. Ann. Intern. Med. 2016, 165, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Barnes, K.G.; Kindrachuk, J.; Lin, A.E.; Wohl, S.; Qu, J.; Tostenson, S.D.; Dorman, W.R.; Busby, M.; Siddle, K.J.; Luo, C.Y.; et al. Evidence of Ebola Virus Replication and High Concentration in Semen of a Patient During Recovery. Clin. Infect. Dis. 2017, 65, 1400–1403. [Google Scholar] [CrossRef] [PubMed]
- Kash, J.C.; Walters, K.A.; Kindrachuk, J.; Baxter, D.; Scherler, K.; Janosko, K.B.; Adams, R.D.; Herbert, A.S.; James, R.M.; Stonier, S.W.; et al. Longitudinal peripheral blood transcriptional analysis of a patient with severe Ebola virus disease. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Rivera, M.M.; McBurney, R.; Park, Y.; Haynes-Williams, V.; Rehermann, B.; Alter, H.J.; Herrine, S.K.; Liang, T.J.; Hoofnagle, J.H.; et al. The natural history of acute hepatitis C: Clinical presentation, laboratory findings and treatment outcomes. Aliment. Pharmacol. Ther. 2011, 33, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Shteyer, E.; Shekhtman, L.; Zinger, T.; Harari, S.; Gafanovich, I.; Wolf, D.; Ivgi, H.; Barsuk, R.; Dery, I.; Armoni, D.; et al. Modeling suggests that microliter volumes of contaminated blood caused an outbreak of hepatitis C during computerized tomography. PLoS ONE 2019, 14, e0210173. [Google Scholar] [CrossRef]
- Martines, R.B.; Ng, D.L.; Greer, P.W.; Rollin, P.E.; Zaki, S.R. Tissue and cellular tropism, pathology and pathogenesis of Ebola and Marburg viruses. J. Pathol. 2015, 235, 153–174. [Google Scholar] [CrossRef] [PubMed]
- Baseler, L.; Chertow, D.S.; Johnson, K.M.; Feldmann, H.; Morens, D.M. The Pathogenesis of Ebola Virus Disease. Annu. Rev. Pathol. 2017, 12, 387–418. [Google Scholar] [CrossRef] [PubMed]
- Vetter, P.; Fischer, W.A.; Schibler, M.; Jacobs, M.; Bausch, D.G.; Kaiser, L. Ebola Virus Shedding and Transmission: Review of Current Evidence. J. Infect. Dis. 2016, 214, S177–S184. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, R.A.; Pais, S.; Reynolds, T.B.; Kaplowitz, N. Serum alanine aminotransferase in skeletal muscle diseases. Hepatology 2005, 41, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Hunt, L.; Gupta-Wright, A.; Simms, V.; Tamba, F.; Knott, V.; Tamba, K.; Heisenberg-Mansaray, S.; Tamba, E.; Sheriff, A.; Conteh, S.; et al. Clinical presentation, biochemical, and haematological parameters and their association with outcome in patients with Ebola virus disease: An observational cohort study. Lancet Infect. Dis. 2015, 15, 1292–1299. [Google Scholar] [CrossRef]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Cournac, J.M.; Karkowski, L.; Bordes, J.; Aletti, M.; Duron, S.; Janvier, F.; Foissaud, V.; Savini, H.; de Greslan, T.; Rousseau, C.; et al. Rhabdomyolysis in Ebola Virus Disease. Results of an Observational Study in a Treatment Center in Guinea. Clin. Infect. Dis. 2016, 62, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.S.; Huzella, L.M.; Jahrling, P.B.; Bollinger, L.; Olinger, G.G., Jr.; Hensley, L.E. Nonhuman Primate Models of Ebola Virus Disease. Curr. Top. Microbiol. Immunol. 2017, 411, 171–193. [Google Scholar] [CrossRef]
- PREVAIL II Writing Group; Multi-National PREVAIL II Study Team; Davey, R.T., Jr.; Dodd, L.; Proschan, M.A.; Neaton, J.; Neuhaus Nordwall, J.; Koopmeiners, J.S.; Beigel, J.; Tierney, J.; et al. A Randomized, Controlled Trial of ZMapp for Ebola Virus Infection. N. Engl. J. Med. 2016, 375, 1448–1456. [Google Scholar] [CrossRef]
- Mulangu, S.; Dodd, L.E.; Davey, R.T., Jr.; Tshiani Mbaya, O.; Proschan, M.; Mukadi, D.; Lusakibanza Manzo, M.; Nzolo, D.; Tshomba Oloma, A.; Ibanda, A.; et al. A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics. N. Engl. J. Med. 2019. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chertow, D.S.; Shekhtman, L.; Lurie, Y.; Davey, R.T.; Heller, T.; Dahari, H. Modeling Challenges of Ebola Virus–Host Dynamics during Infection and Treatment. Viruses 2020, 12, 106. https://doi.org/10.3390/v12010106
Chertow DS, Shekhtman L, Lurie Y, Davey RT, Heller T, Dahari H. Modeling Challenges of Ebola Virus–Host Dynamics during Infection and Treatment. Viruses. 2020; 12(1):106. https://doi.org/10.3390/v12010106
Chicago/Turabian StyleChertow, Daniel S., Louis Shekhtman, Yoav Lurie, Richard T. Davey, Theo Heller, and Harel Dahari. 2020. "Modeling Challenges of Ebola Virus–Host Dynamics during Infection and Treatment" Viruses 12, no. 1: 106. https://doi.org/10.3390/v12010106
APA StyleChertow, D. S., Shekhtman, L., Lurie, Y., Davey, R. T., Heller, T., & Dahari, H. (2020). Modeling Challenges of Ebola Virus–Host Dynamics during Infection and Treatment. Viruses, 12(1), 106. https://doi.org/10.3390/v12010106