Chasing Intracellular Zika Virus Using Proteomics


Abstract
1. Introduction
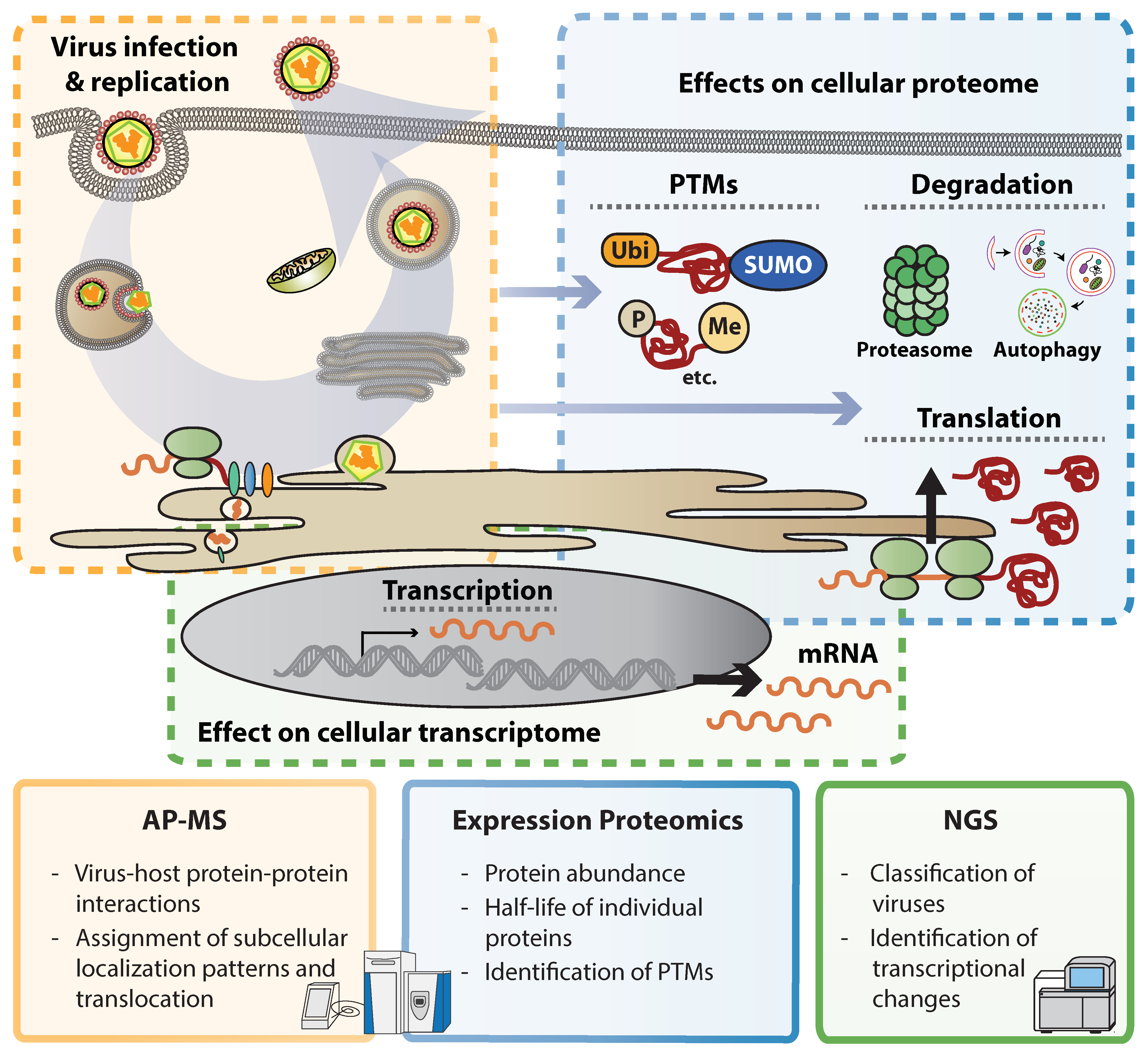
2. Illuminating Dark Matter: Unbiased Approaches to Study Virus–Host Interactions
3. Flavivirus Interactions with the Cellular Proteome
3.1. ZIKV–Host Interactions
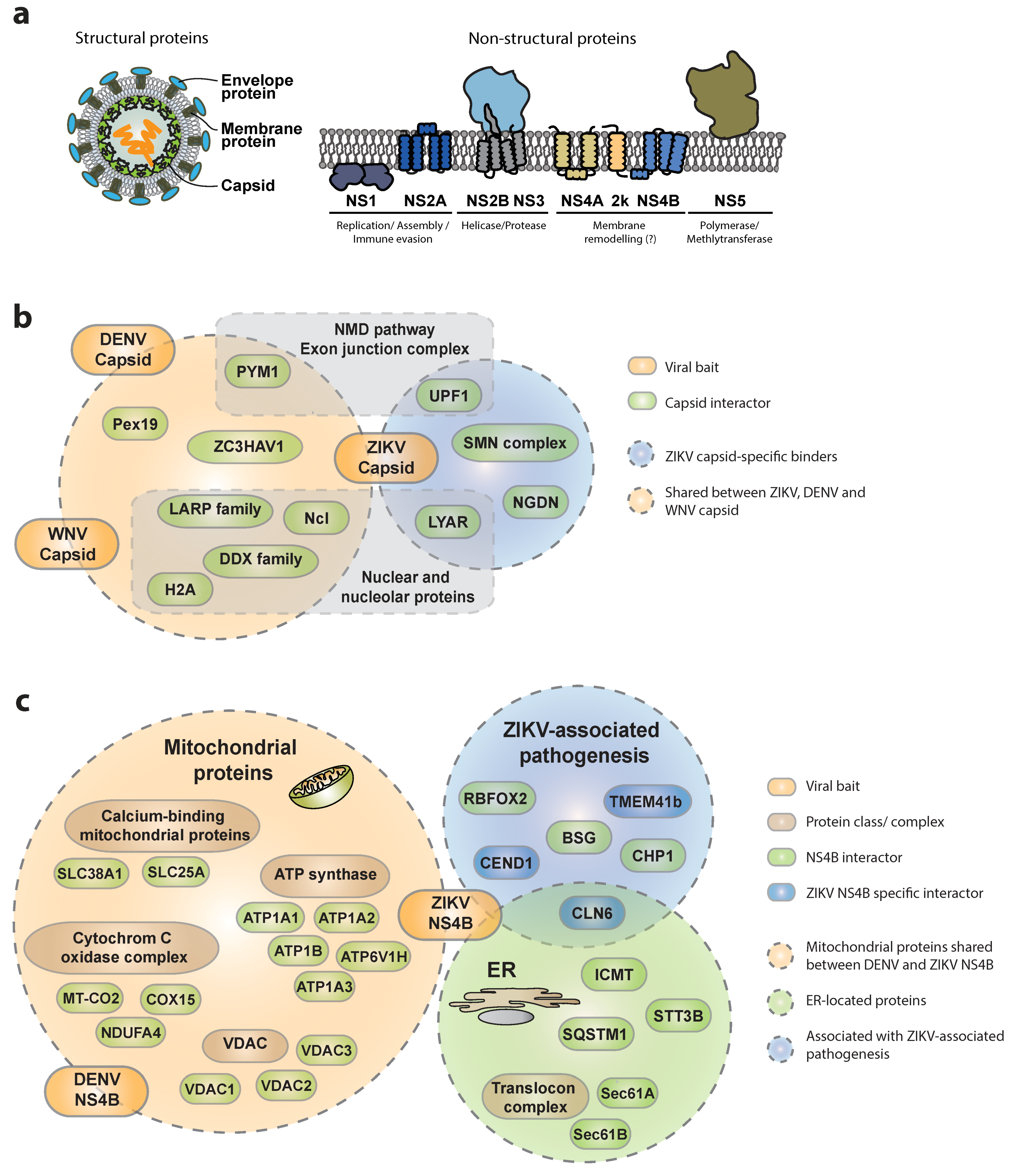
3.1.1. Interactions of ZIKV Capsid with Host Proteins
3.1.2. Interactions of ZIKV NS4B with the Host
3.1.3. Other Viral Proteins
3.1.4. Interactions of ZIKV Viral RNA with the Host
3.2. Global Proteome Expression Affected by ZIKV Infection
3.3. Phosphorylation and Other PTMs Modulated by ZIKV
4. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Lazear, H.M.; Diamond, M.S. Zika Virus: New Clinical Syndromes and Its Emergence in the Western Hemisphere. J. Virol. 2016, 90, 4864–4875. [Google Scholar] [CrossRef] [PubMed]
- Cauchemez, S.; Besnard, M.; Bompard, P.; Dub, T.; Guillemette-Artur, P.; Eyrolle-Guignot, D.; Salje, H.; Van Kerkhove, M.D.; Abadie, V.; Garel, C.; et al. Association between Zika virus and microcephaly in French Polynesia, 2013–2015: A retrospective study. Lancet 2016, 387, 2125–2132. [Google Scholar] [CrossRef]
- Ventura, C.V.; Maia, M.; Bravo-Filho, V.; Gois, A.L.; Belfort, R., Jr. Zika virus in Brazil and macular atrophy in a child with microcephaly. Lancet 2016, 387, 228. [Google Scholar] [CrossRef]
- Ma, W.; Li, S.; Ma, S.; Jia, L.; Zhang, F.; Zhang, Y.; Zhang, J.; Wong, G.; Zhang, S.; Lu, X.; et al. Zika Virus Causes Testis Damage and Leads to Male Infertility in Mice. Cell 2017, 168, 542. [Google Scholar] [CrossRef] [PubMed]
- Kodati, S.; Palmore, T.N.; Spellman, F.A.; Cunningham, D.; Weistrop, B.; Sen, H.N. Bilateral posterior uveitis associated with Zika virus infection. Lancet 2017, 389, 125–126. [Google Scholar] [CrossRef]
- Miner, J.J.; Cao, B.; Govero, J.; Smith, A.M.; Fernandez, E.; Cabrera, O.H.; Garber, C.; Noll, M.; Klein, R.S.; Noguchi, K.K.; et al. Zika Virus Infection during Pregnancy in Mice Causes Placental Damage and Fetal Demise. Cell 2016, 165, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popovic, M.; Poljsak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodusek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 2018, 560, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Kandasamy, K.; Alvisi, G.; Mulhern, O.; Sacco, R.; Habjan, M.; Binder, M.; Stefanovic, A.; Eberle, C.A.; Goncalves, A.; et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 2012, 487, 486–490. [Google Scholar] [CrossRef]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef]
- Batra, J.; Hultquist, J.F.; Liu, D.; Shtanko, O.; Von Dollen, J.; Satkamp, L.; Jang, G.M.; Luthra, P.; Schwarz, T.M.; Small, G.I.; et al. Protein Interaction Mapping Identifies RBBP6 as a Negative Regulator of Ebola Virus Replication. Cell 2018, 175, 1917–1930.e13. [Google Scholar] [CrossRef] [PubMed]
- Davis, Z.H.; Verschueren, E.; Jang, G.M.; Kleffman, K.; Johnson, J.R.; Park, J.; Von Dollen, J.; Maher, M.C.; Johnson, T.; Newton, W.; et al. Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol. Cell 2015, 57, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Dechtawewat, T.; Paemanee, A.; Roytrakul, S.; Songprakhon, P.; Limjindaporn, T.; Yenchitsomanus, P.T.; Saitornuang, S.; Puttikhunt, C.; Kasinrerk, W.; Malasit, P.; et al. Mass spectrometric analysis of host cell proteins interacting with dengue virus nonstructural protein 1 in dengue virus-infected HepG2 cells. Biochim. Biophys. Acta 2016, 1864, 1270–1280. [Google Scholar] [CrossRef] [PubMed]
- Marceau, C.D.; Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Brewer, S.M.; Fuchs, G.; Swaminathan, K.; Mata, M.A.; Elias, J.E.; Sarnow, P.; et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 2016, 535, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Savidis, G.; McDougall, W.M.; Meraner, P.; Perreira, J.M.; Portmann, J.M.; Trincucci, G.; John, S.P.; Aker, A.M.; Renzette, N.; Robbins, D.R.; et al. Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell Rep. 2016, 16, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Sessions, O.M.; Barrows, N.J.; Souza-Neto, J.A.; Robinson, T.J.; Hershey, C.L.; Rodgers, M.A.; Ramirez, J.L.; Dimopoulos, G.; Yang, P.L.; Pearson, J.L.; et al. Discovery of insect and human dengue virus host factors. Nature 2009, 458, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- Dukhovny, A.; Lamkiewicz, K.; Chen, Q.; Fricke, M.; Jabrane-Ferrat, N.; Marz, M.; Jung, J.U.; Sklan, E.H. A CRISPR Activation Screen Identifies Genes That Protect against Zika Virus Infection. J. Virol. 2019, 93, e00211-19. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Muffat, J.; Omer Javed, A.; Keys, H.R.; Lungjangwa, T.; Bosch, I.; Khan, M.; Virgilio, M.C.; Gehrke, L.; Sabatini, D.M.; et al. Genome-wide CRISPR screen for Zika virus resistance in human neural cells. Proc. Natl. Acad. Sci. USA 2019, 116, 9527–9532. [Google Scholar] [CrossRef]
- Ma, H.; Dang, Y.; Wu, Y.; Jia, G.; Anaya, E.; Zhang, J.; Abraham, S.; Choi, J.G.; Shi, G.; Qi, L.; et al. A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death. Cell Rep. 2015, 12, 673–683. [Google Scholar] [CrossRef]
- Richardson, R.B.; Ohlson, M.B.; Eitson, J.L.; Kumar, A.; McDougal, M.B.; Boys, I.N.; Mar, K.B.; De La Cruz-Rivera, P.C.; Douglas, C.; Konopka, G.; et al. A CRISPR screen identifies IFI6 as an ER-resident interferon effector that blocks flavivirus replication. Nat. Microbiol. 2018, 3, 1214–1223. [Google Scholar] [CrossRef]
- Zhang, R.; Miner, J.J.; Gorman, M.J.; Rausch, K.; Ramage, H.; White, J.P.; Zuiani, A.; Zhang, P.; Fernandez, E.; Zhang, Q.; et al. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 2016, 535, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Metsky, H.C.; Matranga, C.B.; Wohl, S.; Schaffner, S.F.; Freije, C.A.; Winnicki, S.M.; West, K.; Qu, J.; Baniecki, M.L.; Gladden-Young, A.; et al. Zika virus evolution and spread in the Americas. Nature 2017, 546, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.G.; Shi, W.F.; Liu, D.; Qian, J.; Liang, L.; Bo, X.C.; Liu, J.; Ren, H.G.; Fan, H.; Ni, M.; et al. China Mobile Laboratory Testing Team in Sierra, L. Genetic diversity and evolutionary dynamics of Ebola virus in Sierra Leone. Nature 2015, 524, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Edfors, F.; Danielsson, F.; Hallstrom, B.M.; Kall, L.; Lundberg, E.; Ponten, F.; Forsstrom, B.; Uhlen, M. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol. Syst. Biol. 2016, 12, 883. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Itzhak, D.N.; Tyanova, S.; Cox, J.; Borner, G.H. Global, quantitative and dynamic mapping of protein subcellular localization. Elife 2016, 5, e16950. [Google Scholar] [CrossRef] [PubMed]
- Jean Beltran, P.M.; Mathias, R.A.; Cristea, I.M. A Portrait of the Human Organelle Proteome in Space and Time during Cytomegalovirus Infection. Cell Syst. 2016, 3, 361–373.e6. [Google Scholar] [CrossRef]
- Sinitcyn, P.; Rudolph, J.D.; Cox, J. Computational Methods for Understanding Mass Spectrometry–Based Shotgun Proteomics Data. Annu. Rev. Biomed. Data Sci. 2018, 1, 207–234. [Google Scholar] [CrossRef]
- Taguwa, S.; Maringer, K.; Li, X.; Bernal-Rubio, D.; Rauch, J.N.; Gestwicki, J.E.; Andino, R.; Fernandez-Sesma, A.; Frydman, J. Defining Hsp70 Subnetworks in Dengue Virus Replication Reveals Key Vulnerability in Flavivirus Infection. Cell 2015, 163, 1108–1123. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Rebsamen, M.; Kandasamy, R.K.; Superti-Furga, G. Protein interaction networks in innate immunity. Trends Immunol. 2013, 34, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Scaturro, P.; Stukalov, A.; Haas, D.A.; Cortese, M.; Draganova, K.; Plaszczyca, A.; Bartenschlager, R.; Gotz, M.; Pichlmair, A. An orthogonal proteomic survey uncovers novel Zika virus host factors. Nature 2018, 561, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Balinsky, C.A.; Schmeisser, H.; Ganesan, S.; Singh, K.; Pierson, T.C.; Zoon, K.C. Nucleolin interacts with the dengue virus capsid protein and plays a role in formation of infectious virus particles. J. Virol. 2013, 87, 13094–13106. [Google Scholar] [CrossRef] [PubMed]
- Colpitts, T.M.; Barthel, S.; Wang, P.; Fikrig, E. Dengue virus capsid protein binds core histones and inhibits nucleosome formation in human liver cells. PLoS ONE 2011, 6, e24365. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Hou, S.; Malik-Soni, N.; Xu, Z.; Kumar, A.; Rachubinski, R.A.; Frappier, L.; Hobman, T.C. Flavivirus Infection Impairs Peroxisome Biogenesis and Early Antiviral Signaling. J. Virol. 2015, 89, 12349–12361. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, B.; Yang, A.; Lu, R.; Wang, W.; Zhou, Y.; Shi, G.; Kwon, S.W.; Zhao, Y.; Jin, Y. Ly-1 antibody reactive clone is an important nucleolar protein for control of self-renewal and differentiation in embryonic stem cells. Stem Cells 2009, 27, 1244–1254. [Google Scholar] [CrossRef]
- Yang, C.; Liu, X.; Gao, Q.; Cheng, T.; Xiao, R.; Ming, F.; Zhang, S.; Jin, M.; Chen, H.; Ma, W.; et al. The Nucleolar Protein LYAR Facilitates Ribonucleoprotein Assembly of Influenza a Virus. J. Virol. 2018, 92, e01042-18. [Google Scholar] [CrossRef]
- Coyaud, E.; Ranadheera, C.; Cheng, D.; Goncalves, J.; Dyakov, B.J.A.; Laurent, E.M.N.; St-Germain, J.; Pelletier, L.; Gingras, A.C.; Brumell, J.H.; et al. Global Interactomics Uncovers Extensive Organellar Targeting by Zika Virus. Mol. Cell Proteom. 2018, 17, 2242–2255. [Google Scholar] [CrossRef]
- Shah, P.S.; Link, N.; Jang, G.M.; Sharp, P.P.; Zhu, T.; Swaney, D.L.; Johnson, J.R.; Von Dollen, J.; Ramage, H.R.; Satkamp, L.; et al. Comparative Flavivirus-Host Protein Interaction Mapping Reveals Mechanisms of Dengue and Zika Virus Pathogenesis. Cell 2018, 175, 1931–1945.e18. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Johnson, J.R.; Truong, B.; Kim, G.; Weinbren, N.; Dittmar, M.; Shah, P.S.; Von Dollen, J.; Newton, B.W.; Jang, G.M.; et al. Identification of antiviral roles for the exon-junction complex and nonsense-mediated decay in flaviviral infection. Nat. Microbiol. 2019, 4, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.A.; Goncalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Lau, Z.; Cheung, P.; Aguilar, E.G.; Schneider, W.M.; Bozzacco, L.; Molina, H.; Buehler, E.; Takaoka, A.; Rice, C.M.; et al. TRIM25 Enhances the Antiviral Action of Zinc-Finger Antiviral Protein (ZAP). PLoS Pathog. 2017, 13, e1006145. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.P.; Chiu, H.; Yang, C.F.; Lee, Y.L.; Chiu, F.L.; Kuo, H.C.; Lin, R.J.; Lin, Y.L. Inhibition of Japanese encephalitis virus infection by the host zinc-finger antiviral protein. PLoS Pathog. 2018, 14, e1007166. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Leon, K.E.; Khalid, M.M.; Tomar, S.; Jimenez-Morales, D.; Dunlap, M.; Kaye, J.A.; Shah, P.S.; Finkbeiner, S.; Krogan, N.J.; et al. The Cellular NMD Pathway Restricts Zika Virus Infection and Is Targeted by the Viral Capsid Protein. MBio 2018, 9, e02126-18. [Google Scholar] [CrossRef] [PubMed]
- Chatel-Chaix, L.; Cortese, M.; Romero-Brey, I.; Bender, S.; Neufeldt, C.J.; Fischl, W.; Scaturro, P.; Schieber, N.; Schwab, Y.; Fischer, B.; et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host Microbe. 2016, 20, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Metz, P.; Chiramel, A.; Chatel-Chaix, L.; Alvisi, G.; Bankhead, P.; Mora-Rodriguez, R.; Long, G.; Hamacher-Brady, A.; Brady, N.R.; Bartenschlager, R. Dengue Virus Inhibition of Autophagic Flux and Dependency of Viral Replication on Proteasomal Degradation of the Autophagy Receptor p62. J. Virol. 2015, 89, 8026–8041. [Google Scholar] [CrossRef]
- Heaton, N.S.; Randall, G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef]
- Liu, Y.; Cherry, S. Zika virus infection activates sting-dependent antiviral autophagy in the Drosophila brain. Autophagy 2019, 15, 174–175. [Google Scholar] [CrossRef]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.S.; Lee, S.A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Moshkina, N.; Fenouil, R.; Gardner, T.J.; Aguirre, S.; Shah, P.S.; Zhao, N.; Manganaro, L.; Hultquist, J.F.; Noel, J.; et al. Targeting Viral Proteostasis Limits Influenza Virus, HIV, and Dengue Virus Infection. Immunity 2016, 44, 46–58. [Google Scholar] [CrossRef] [PubMed]
- De Maio, F.A.; Risso, G.; Iglesias, N.G.; Shah, P.; Pozzi, B.; Gebhard, L.G.; Mammi, P.; Mancini, E.; Yanovsky, M.J.; Andino, R.; et al. The Dengue Virus NS5 Protein Intrudes in the Cellular Spliceosome and Modulates Splicing. PLoS Pathog. 2016, 12, e1005841. [Google Scholar] [CrossRef] [PubMed]
- Kovanich, D.; Saisawang, C.; Sittipaisankul, P.; Ramphan, S.; Kalpongnukul, N.; Somparn, P.; Pisitkun, T.; Smith, D.R. Analysis of the Zika and Japanese Encephalitis Virus NS5 Interactomes. J. Proteome. Res. 2019, 18, 3203–3218. [Google Scholar] [CrossRef] [PubMed]
- Hafirassou, M.L.; Meertens, L.; Umana-Diaz, C.; Labeau, A.; Dejarnac, O.; Bonnet-Madin, L.; Kummerer, B.M.; Delaugerre, C.; Roingeard, P.; Vidalain, P.O.; et al. A Global Interactome Map of the Dengue Virus NS1 Identifies Virus Restriction and Dependency Host Factors. Cell Rep. 2017, 21, 3900–3913. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.L.; Inoue, T.; Chen, Y.J.; Chang, A.; Tsai, B.; Tai, A.W. The ER Membrane Protein Complex Promotes Biogenesis of Dengue and Zika Virus Non-structural Multi-pass Transmembrane Proteins to Support Infection. Cell Rep. 2019, 27, 1666–1674.e4. [Google Scholar] [CrossRef] [PubMed]
- Barrows, N.J.; Campos, R.K.; Powell, S.T.; Prasanth, K.R.; Schott-Lerner, G.; Soto-Acosta, R.; Galarza-Munoz, G.; McGrath, E.L.; Urrabaz-Garza, R.; Gao, J.; et al. A Screen of FDA-Approved Drugs for Inhibitors of Zika Virus Infection. Cell Host Microbe 2016, 20, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Youn, S.; Li, T.; McCune, B.T.; Edeling, M.A.; Fremont, D.H.; Cristea, I.M.; Diamond, M.S. Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J. Virol. 2012, 86, 7360–7371. [Google Scholar] [CrossRef]
- Plaszczyca, A.; Scaturro, P.; Neufeldt, C.J.; Cortese, M.; Cerikan, B.; Ferla, S.; Brancale, A.; Pichlmair, A.; Bartenschlager, R. A novel interaction between dengue virus nonstructural protein 1 and the NS4A-2K-4B precursor is required for viral RNA replication but not for formation of the membranous replication organelle. PLoS Pathog. 2019, 15, e1007736. [Google Scholar] [CrossRef] [PubMed]
- Chatel-Chaix, L.; Fischl, W.; Scaturro, P.; Cortese, M.; Kallis, S.; Bartenschlager, M.; Fischer, B.; Bartenschlager, R. A Combined Genetic-Proteomic Approach Identifies Residues within Dengue Virus NS4B Critical for Interaction with NS3 and Viral Replication. J. Virol. 2015, 89, 7170–7186. [Google Scholar] [CrossRef]
- Roby, J.A.; Pijlman, G.P.; Wilusz, J.; Khromykh, A.A. Noncoding subgenomic flavivirus RNA: Multiple functions in West Nile virus pathogenesis and modulation of host responses. Viruses 2014, 6, 404–427. [Google Scholar] [CrossRef] [PubMed]
- Chavali, P.L.; Stojic, L.; Meredith, L.W.; Joseph, N.; Nahorski, M.S.; Sanford, T.J.; Sweeney, T.R.; Krishna, B.A.; Hosmillo, M.; Firth, A.E.; et al. Neurodevelopmental protein Musashi-1 interacts with the Zika genome and promotes viral replication. Science 2017, 357, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Charley, P.A.; Wilusz, J. Sponging of cellular proteins by viral RNAs. Curr. Opin. Virol. 2014, 9, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Viktorovskaya, O.V.; Greco, T.M.; Cristea, I.M.; Thompson, S.R. Identification of RNA Binding Proteins Associated with Dengue Virus RNA in Infected Cells Reveals Temporally Distinct Host Factor Requirements. PLoS Negl. Trop. Dis. 2016, 10, e0004921. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.L.; Soderblom, E.J.; Bradrick, S.S.; Garcia-Blanco, M.A. Identification of Proteins Bound to Dengue Viral RNA in Vivo Reveals New Host Proteins Important for Virus Replication. MBio 2016, 7, e01865-e15. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Zhang, Q.C.; da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.; Heard, E.; Chang, H.Y. Systematic discovery of Xist RNA binding proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Ooi, Y.S.; Majzoub, K.; Flynn, R.A.; Mata, M.A.; Diep, J.; Li, J.K.; van Buuren, N.; Rumachik, N.; Johnson, A.G.; Puschnik, A.S.; et al. An RNA-centric dissection of host complexes controlling flavivirus infection. Nat. Microbiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.C.; Hannemann, H.; Heesom, K.J.; Matthews, D.A.; Davidson, A.D. High-throughput quantitative proteomic analysis of dengue virus type 2 infected A549 cells. PLoS ONE 2014, 9, e93305. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, B.; Boucomont-Chapeaublanc, E.; Peyrefitte, C.N.; Belghazi, M.; Fusai, T.; Rogier, C.; Tolou, H.J.; Almeras, L. Identification of cellular proteome modifications in response to West Nile virus infection. Mol. Cell Proteom. 2009, 8, 1623–1637. [Google Scholar] [CrossRef] [PubMed]
- Garcez, P.P.; Nascimento, J.M.; de Vasconcelos, J.M.; Madeiro da Costa, R.; Delvecchio, R.; Trindade, P.; Loiola, E.C.; Higa, L.M.; Cassoli, J.S.; Vitoria, G.; et al. Zika virus disrupts molecular fingerprinting of human neurospheres. Sci. Rep. 2017, 7, 40780. [Google Scholar] [CrossRef] [PubMed]
- Hammack, C.; Ogden, S.C.; Madden, J.C., Jr.; Medina, A.; Xu, C.; Phillips, E.; Son, Y.; Cone, A.; Giovinazzi, S.; Didier, R.A.; et al. Zika virus infection induces DNA damage response in human neural progenitors that enhances viral replication. J. Virol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Beys-da-Silva, W.O.; Rosa, R.L.; Santi, L.; Berger, M.; Park, S.K.; Campos, A.R.; Terraciano, P.; Varela, A.P.M.; Teixeira, T.F.; Roehe, P.M.; et al. Zika Virus Infection of Human Mesenchymal Stem Cells Promotes Differential Expression of Proteins Linked to Several Neurological Diseases. Mol. Neurobiol. 2019, 56, 4708–4717. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Fernandes, L.; Cugola, F.R.; Russo, F.B.; Kawahara, R.; de Melo Freire, C.C.; Leite, P.E.C.; Bassi Stern, A.C.; Angeli, C.B.; de Oliveira, D.B.L.; Melo, S.R.; et al. Zika Virus Impairs Neurogenesis and Synaptogenesis Pathways in Human Neural Stem Cells and Neurons. Front. Cell Neurosci. 2019, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhang, H.; He, W.; Zhu, B.; Zhou, D.; Chen, Z.; Ashraf, U.; Wei, Y.; Liu, Z.; Fu, Z.F.; et al. Quantitative phosphoproteomic analysis identifies the critical role of JNK1 in neuroinflammation induced by Japanese encephalitis virus. Sci. Signal 2016, 9, ra98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, J.; Ye, J.; Ashraf, U.; Chen, Z.; Zhu, B.; He, W.; Xu, Q.; Wei, Y.; Chen, H.; et al. Quantitative Label-Free Phosphoproteomics Reveals Differentially Regulated Protein Phosphorylation Involved in West Nile Virus-Induced Host Inflammatory Response. J. Proteome Res. 2015, 14, 5157–5168. [Google Scholar] [CrossRef]
- Souza, B.S.; Sampaio, G.L.; Pereira, C.S.; Campos, G.S.; Sardi, S.I.; Freitas, L.A.; Figueira, C.P.; Paredes, B.D.; Nonaka, C.K.; Azevedo, C.M.; et al. Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci. Rep. 2016, 6, 39775. [Google Scholar] [CrossRef] [PubMed]
- Ghouzzi, V.E.; Bianchi, F.T.; Molineris, I.; Mounce, B.C.; Berto, G.E.; Rak, M.; Lebon, S.; Aubry, L.; Tocco, C.; Gai, M.; et al. ZIKA virus elicits P53 activation and genotoxic stress in human neural progenitors similar to mutations involved in severe forms of genetic microcephaly. Cell Death Dis. 2016, 7, e2440. [Google Scholar] [CrossRef] [PubMed]
- Morooka, T.; Nishida, E. Requirement of p38 mitogen-activated protein kinase for neuronal differentiation in PC12 cells. J. Biol. Chem. 1998, 273, 24285–24288. [Google Scholar] [CrossRef]
- Xu, X.H.; Deng, C.Y.; Liu, Y.; He, M.; Peng, J.; Wang, T.; Yuan, L.; Zheng, Z.S.; Blackshear, P.J.; Luo, Z.G. MARCKS regulates membrane targeting of Rab10 vesicles to promote axon development. Cell Res. 2014, 24, 576–594. [Google Scholar] [CrossRef] [PubMed]
- Morimura, R.; Nozawa, K.; Tanaka, H.; Ohshima, T. Phosphorylation of Dpsyl2 (CRMP2) and Dpsyl3 (CRMP4) is required for positioning of caudal primary motor neurons in the zebrafish spinal cord. Dev. Neurobiol. 2013, 73, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lan, Y.; Li, M.Y.; Lamers, M.M.; Fusade-Boyer, M.; Klemm, E.; Thiele, C.; Ashour, J.; Sanyal, S. Flaviviruses Exploit the Lipid Droplet Protein AUP1 to Trigger Lipophagy and Drive Virus Production. Cell Host Microbe 2018, 23, 819–831.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kuster, B. Proteomics Is Not an Island: Multi-omics Integration Is the Key to Understanding Biological Systems. Mol. Cell Proteom. 2019, 18 (Suppl. 1), S1–S4. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene Name | Protein Name | Gene Name | Protein Name |
|---|---|---|---|
| AKT | RAC-alpha serine/threonine-protein kinase | ICMT | protein-S-isoprenylcysteine O-methyltransferase |
| ANKLE2 | Ankyrin repeat and LEM domain-containing protein 2 | LARP | La-related protein |
| ANKRD17 | ankyrin repeat domain-containing protein 17 | LEO1 | RNA polymerase-associated protein 1 |
| ATF7 | cyclic AMP-dependent transcription factor ATF-7 | LYAR | cell growth-regulating nucleolar protein |
| AUP1 | ancient ubiquitous protein 1 | MAP4K4 | mitogen-activated protein kinase kinase kinase kinase 4 |
| BAG3 | BAG family molecular chaperone regulator 3 | MARCKS | myristoylated alanine-rich C-kinase substrate |
| BAZ1B | tyrosine-protein kinase BAZ1B | MRE11A | double-strand break repair protein MRE11 |
| BRCA1 | breast cancer type 1 susceptibility protein | MT-CO2 | cytochrome c oxidaes subunit 2 |
| CD2BP2 | CD2 antigen cytoplasmic tail-binding protein 2 | mTOR | mechanistic target of rapamycin |
| CDC73 | cell division control protein 73 | NCL | nucleolin |
| CDK1 | cyclin-dependent kinase 1 | NDUFA4 | cytochrome c oxidase subunit NDUFA4 |
| CDK2 | cyclin-dependent kinase 2 | NeuroD1 | neurogenic differentiation factor 1 |
| CEND1 | cell-cycle exit and neuronal differentiation protein 1 | NGDN | neuroguidin |
| CLN6 | ceroid-lipofuscinosis neuronal protein 6 | NONO | non-POU domain-containing octamer-binding protein |
| COX15 | cytochrome c oxidase assembly protein COX15 homolog | PAF1 | RNA polymerase II-associated factor homolog |
| CPEB | cytoplasmic polyadenylation element binding protein | PEX19 | peroxisomal biogensis factor 19 |
| CSDE1 | cold-shock domain-containing protein E1 | PYM1 | partner of Y14 and mago |
| CTR9 | RNA polymerase-associated protein CTR9 homolog | RBM8A | RNA-binding protein 8A |
| DCP1-DCP2 | mRNA-decapping enzyme subunit 1-2rbm8A | RBMX | RNA-binding motif protein X chromosome |
| DCX | neuronal migration protein doublecortin | RPN2 | dolichyl-diphosphooligosaccharide-protein glycosyltransferase subunit 2 |
| DDOST | dolichyl-diphosphooligosaccharide-protein glycosyltransferase 48 kDa subunit | RRBP1 | ribosome-binding protein 1 |
| DDX | ATP-dependent RNA helicase | SMN1 | survival motor neuron protein 1 |
| DNMT1 | DNA (cytosine-5)-methyltransferase 1 | STT3A/B | dolichyl-diphosphooligosaccharide--protein glycosyltransferase subunit A/B |
| DOK3 | docking protein 3 | TBC1D4 | TBC1 domain family member 4 |
| DPYSL2 | dihydropyrimidinase-related protein 2 | TLR4 | toll-like receptor 4 |
| EEF2K | eukaryotic elongation factor 2 kinase | TMEM41b | transmembrane protein 41beta |
| ERBB2 | Receptor tyrosine-protein kinase erbB-2 | TOP2B | DNA topoisomerase 2-beta |
| FANCD1 | breast cancer type 2 susceptibility protein | TWIST | twist-related protein 1 |
| HDAC7 | histone deacetylase 7 | UPF1 | regulator of nonsense transcripts 1 |
| HMCES | embryonic stem cell-specific 5-hydroxymethylcytosine-binding protein | VDAC1, 2, 3 | voltage-dependent anion selective channel protein 1, 2, 3 |
| hnRNPC | heterogeneous nuclear ribonucleoproteins C1/C2 | WNK1 | Serine/threonine-protein kinase WNK1 |
| HSPB1 | heat shock protein beta-1 | ZC3HAV1 | zinc finger CCCH-type antiviral protein 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scaturro, P.; Kastner, A.L.; Pichlmair, A. Chasing Intracellular Zika Virus Using Proteomics. Viruses 2019, 11, 878. https://doi.org/10.3390/v11090878
Scaturro P, Kastner AL, Pichlmair A. Chasing Intracellular Zika Virus Using Proteomics. Viruses. 2019; 11(9):878. https://doi.org/10.3390/v11090878
Chicago/Turabian StyleScaturro, Pietro, Anna Lena Kastner, and Andreas Pichlmair. 2019. "Chasing Intracellular Zika Virus Using Proteomics" Viruses 11, no. 9: 878. https://doi.org/10.3390/v11090878
APA StyleScaturro, P., Kastner, A. L., & Pichlmair, A. (2019). Chasing Intracellular Zika Virus Using Proteomics. Viruses, 11(9), 878. https://doi.org/10.3390/v11090878