1. Introduction
Rift Valley fever (RVF) is a mosquito-borne anthropozoonosis caused by the RVF virus (RVFV), which belongs to the family
Phenuiviridae in the order of
Bunyavirales [
1]. RVFV is a negative-sense single-stranded RNA virus with a segmented genome, comprising large (L), medium (M) and small (S) segments. The virus mainly circulates in a mosquito-ruminant transmission cycle, and other small mammals, such as rodents, may potentially participate in the maintenance of the virus [
2]. Sheep are most susceptible to infection, manifesting with abortion of pregnant animals and acute mortality among newborns. Humans can become infected via contact with tissues and fluids released during the slaughtering of infected animals or via bites of infected mosquitoes [
3]. Infected humans usually remain asymptomatic or develop a self-limiting febrile illness. However, in some cases, patients develop severe complications manifesting with acute hepatitis, encephalitis or hemorrhagic fever leading to death [
4]. The demonstrated ability to cause large transboundary outbreaks explains the need for a safe and effective vaccine or antiviral therapy. Consequently, the World Health Organization has included RVF on the Blueprint list of priority diseases likely to cause future epidemics for which countermeasures are urgently needed (
http://www.who.int/blueprint/priority-diseases/en/).
Since the virus was first isolated in Kenya in 1930 [
5,
6], subsequent outbreaks occurred in surrounding countries, after which the virus spread to South-, North- and West Africa and the Arabian Peninsula [
7,
8,
9]. The first imported case of RVF in China was reported in 2016, when a Chinese worker returning from Angola was diagnosed with RVFV infection. The virus was isolated from the serum of the patient and was named the BJ01 strain [
10]. Phylogenetic analysis revealed that the imported virus is a reassortant containing the L and M genes from lineage E and the S segment from lineage A [
11]. This imported case underscores the risk for future emergence of RVFV in China and calls for preparedness programs.
The major virulence factor of RVFV is a non-structural 31-kDa protein named NSs that is encoded by the S segment. NSs localizes both in the cytoplasm and nucleus of infected cells, and it forms nuclear filamentous structures through homo-oligomerization [
12]. NSs suppresses host innate immune responses through three strategies, including (i) inhibiting the type I interferon (IFN) system by stabilizing a repressor complex on the interferon-β (IFN-β) promoter [
13]; (ii) dampening antiviral responses by targeting the RNA-dependent protein kinase (PKR) for degradation [
14]; and (iii) inducing a host cellular transcription shut-off by disrupting the assembly of the RNA polymerase II transcription factor II H (TFIIH) complex [
15]. Although various biological functions of NSs have been elucidated, it has remained unclear if NSs filament formation is a determinant of viral virulence. A recent study resolved the crystal structure of a truncated form of NSs (83–248 AA) of the attenuated MP-12 strain and, based on the structure, identified mutations in the fibril interfaces of NSs that abolish its nuclear filament formation (namly T1 and T3) [
16]. However, the virulence of these NSs mutants with compromised NSs nuclear filament formation has not been investigated.
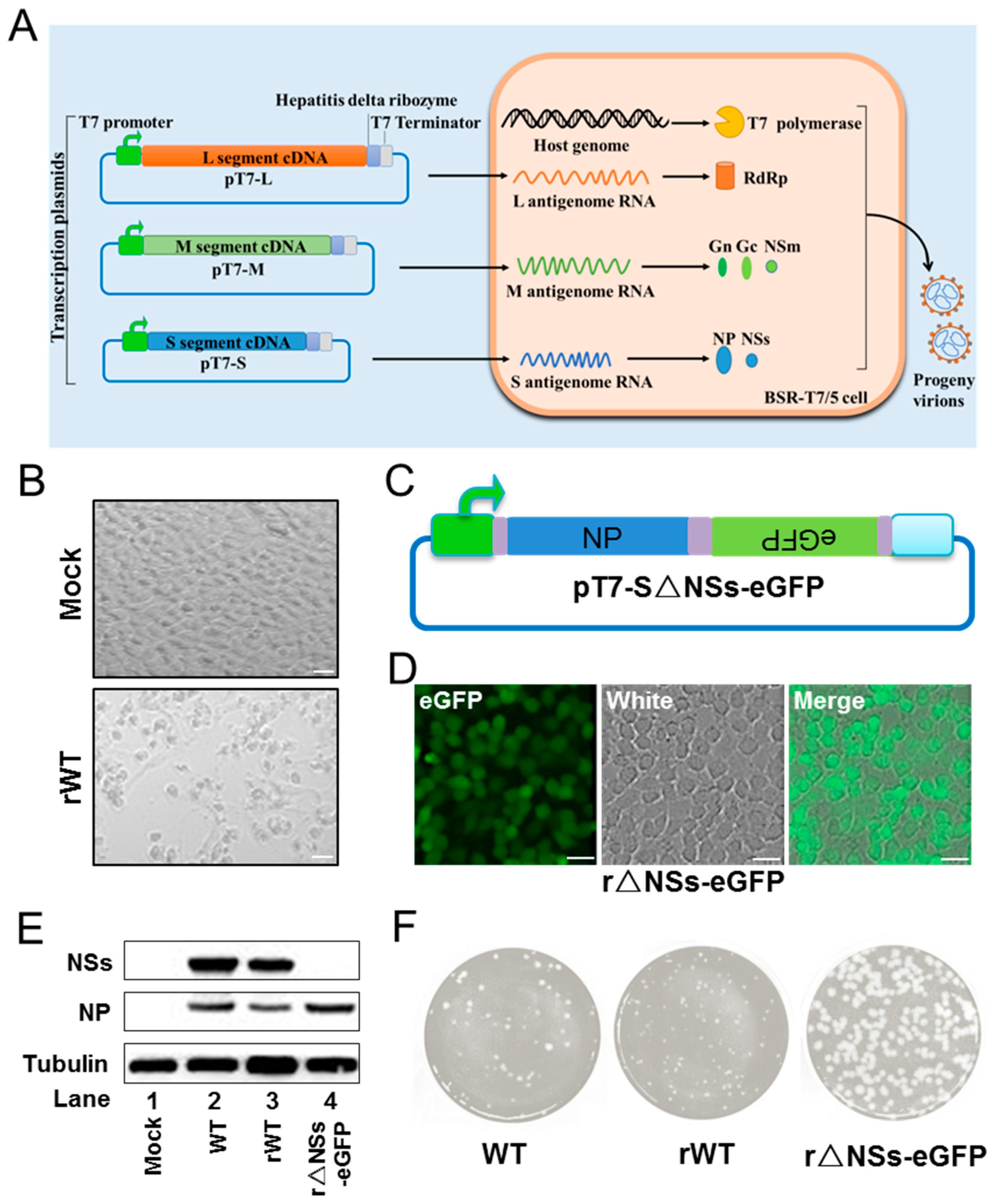
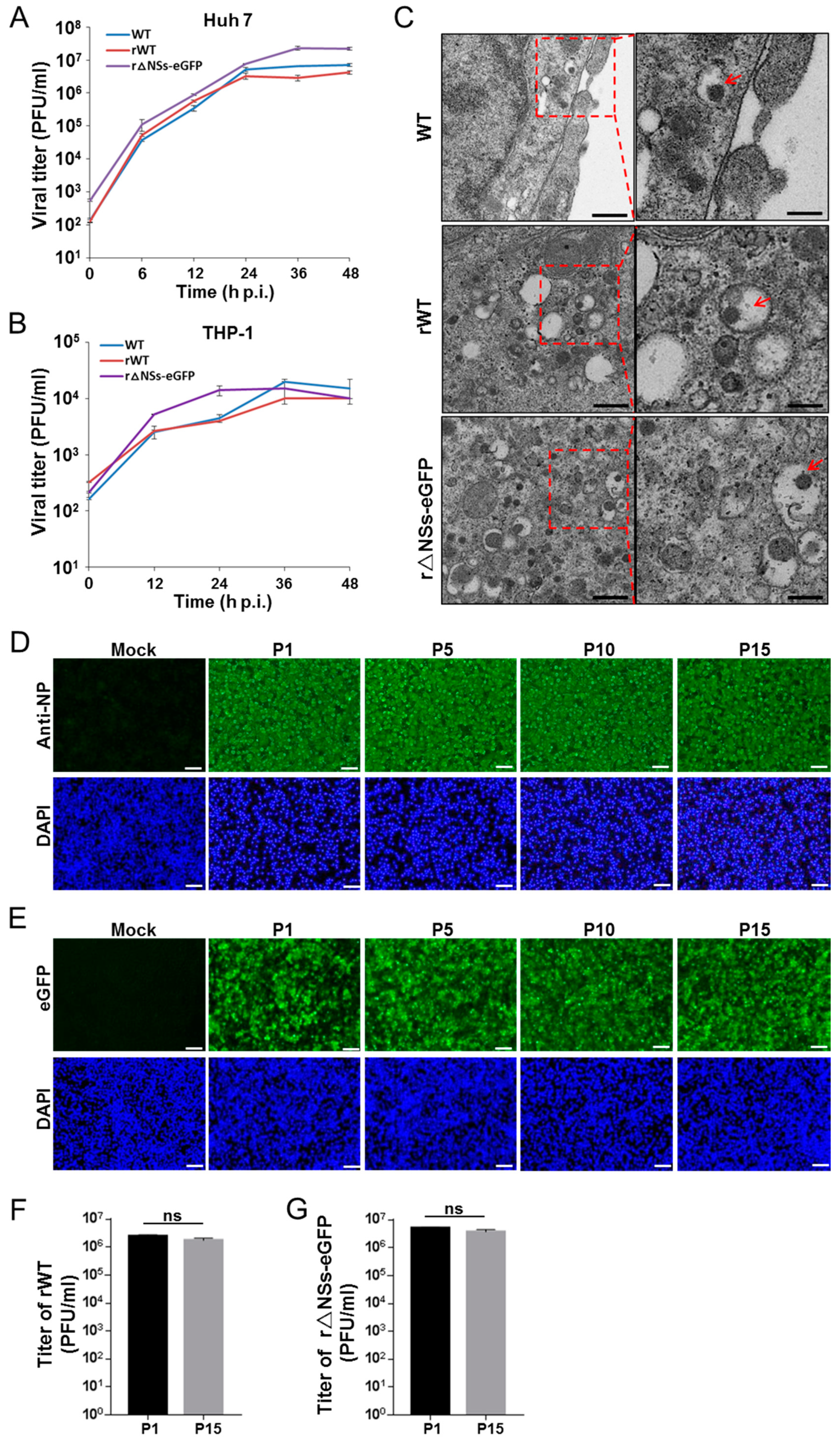
In this study, we generated a T7-based reverse genetics system to rescue the imported BJ01 strain. Both wild-type (WT) and mutant viruses lacking the NSs gene (r△NSs-eGFP) were recovered using this system. The replication properties and pathogenicity of the rescued viruses in vitro and in vivo were investigated. The recombinant BJ01 strain caused a severe cytopathic effect (CPE) in cell cultures and was highly pathogenic for BALB/c mice. To investigate the role of NSs filament formation in virulence, mutations previously shown to compromise NSs nuclear filament formation were introduced into the system. The phenotypes of the mutations were subsequently investigated in vitro and in vivo. The results showed that NSs filament formation is dispensible for efficient replication in vitro and appears important but insufficient for virulence in vivo.
2. Materials and Methods
2.1. Ethics Statement
Animal experiments were performed in agreement with Regulations for the Administration of Affairs Concerning Experimental Animals in China. The procedures were approved by the Laboratory Animal Care and Use Committee of the Wuhan Institute of Virology, Chinese Academy of Sciences (Wuhan, China) on 10 June 2019. The approval code was WIVA38201901.
2.2. Cells and Viruses
Baby hamster kidney (BHK-21), Huh7 and Vero cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% antibiotics (Gibco). BSR-T7/5 cells, stably expressing T7 RNA polymerase, were maintained DMEM supplemented with 10% fetal bovine serum and 1% antibiotics and additionally provided with G418. THP-1 cells were obtained from ATCC, and cultured in RPMI-1640 medium (Gibco) containing 10% FBS. All cell lines were grown in a humidified atmosphere of 5% CO2 at 37 °C. RVFV strains were propagated in BHK-21 cells under BSL-3 conditions, and viral titers were determined by plaque assay in Vero cells.
2.3. Plasmids
The pVSV-T7 plasmid, which contains a T7 promoter and terminator, was a gift from Prof. Gengfu Xiao at the Wuhan Institute of Virology. The cDNAs corresponding to the L, M and S segments of BJ01 were synthesized by reverse transcription-PCR (RT-PCR) from vRNA extracted from the supernatant of infected cells using a QIAamp viral RNA Mini Kit (Qiagen, Dusseldorf, Germany). The PCR products of antigenomes were cloned into pVSV-T7 between the T7 RNA polymerase promoter and terminator with the ClonExpress Ultra One Step Cloning Kit (Vazyme, Nanjing, China), resulting in T7 polymerase-driven plasmids (pT7-L, pT7-M and pT7-S). The NSs deletion and mutations in the oligomer interfaces (T1, T3 and T4) of NSs were constructed through homologous recombination using the plasmid pT7-S as a template.
2.4. Rescue of Recombinant Viruses
Recombinant WT (rWT) virus was rescued by transfecting monolayers of BSR-T7/5 cells (5 × 105) with pT7-L, pT7-M and pT7-S, all of which were 1 μg with Lipofectamine 3000 (Life Technologies, Waltham, MA, USA). After 5 days, an obvious cytopathic effect was observed, and the supernatants were collected and passaged in BHK-21 cells.
The NSs deletion virus (r△NSs-eGFP) was recovered by co-transfecting BSR-T7/5 with pT7-L, pT7-M and pT7-S△NSs-eGFP as described above.
The mutants (rT1, rT3 and rT4) were rescued by co-transfection with pT7-L, pT7-M and pT7-S with T1, T3 or T4 mutation in NSs.
2.5. Plaque Assay
Confluent Vero monolayers were infected with 10-fold dilutions of virus and incubated at 37 °C for 1 h. After absorption, culture medium was replaced by DMEM containing 2% FBS and 1.1% carboxymethyl-cellulose. After 7 days of incubation, the overlay was removed, and cells were stained with 1% crystal violet after fixing with 3.6% formaldehyde.
2.6. Western Blot Analysis
BHK-21 and Huh7 cells infected with indicated viruses were lysed with buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP40, 5 mM EDTA and 10% glycerol). Cell lysates were subjected to 12% SDS-polyacrylamide gel electrophoresis (PAGE) then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Darmstadt, Germany). Proteins were incubated with primary antibodies against nucleocapsid protein (NP), NSs and Tubulin (Abclonal, Wuhan, China), then secondary horseradish peroxidase-conjugated goat anti-rabbit IgG. Protein bands were detected by an enhanced chemiluminescence (ECL) kit (Millipore) using a Chemiluminescence Analyzer (Chemiscope600pro).
2.7. Electron Microscopy (EM) of Infected Cells
Huh7 cells (2 × 106) were infected with RVFV WT (BJ01), rWT or rescued virus lacking the NSs gene (r△NSs-eGFP) at an MOI of 1 for 24 h. Cells were washed three times with PBS, fixed with 2.5% (w/v) glutaraldehyde in 0.1 M sodium chloride and and processed for Electron Microscopy (EM). The viral particles were observed under a transmission electron microscope (FEI Tecnai G2 microscope at 200 kV).
2.8. Fluorescence and Immunofluorescence Microscopy
BHK-21 cells infected with r△NSs-eGFP were fixed with 4% paraformaldehyde (PFA) and incubated with 4′,6-diamidino-2-phenylindole (DAPI). BHK-21 cells infected with rWT were fixed and permeabilized with 0.2% (vol/vol) Triton X-100, then blocked with PBS containing 3% bovine serum albumin (BSA). Cells were incubated with anti-NP antibody for 1 h at room temperature (RT), followed by incubation with Alexa Fluor 488-conjugated goat anti-rabbit IgG, then washed and incubated with DAPI. Images were captured by an epifluorescence microscope (Olympus IX73).
For the immunostaining of NSs, Vero and Huh7 cells were mock treated or infected with rWT and mutants (rT1, rT3 and rT4) at an MOI of 5. At 24 h p.i., cells were fixed and permeabilized as described above, then blocked with 3% BSA. Cells were incubated with anti-NSs antibody for 1 h at RT, followed by incubation with Alexa Fluor 561-conjugated goat anti-rabbit IgG. After that, cells were incubated with DAPI, and analyzed using a confocal microscope (Andor Dragonfly 202).
2.9. Determination of In Vivo Virulence of Mutant Viruses
BALB/c mice were purchased from Charles River Laboratories (Beijing, China). Sixty 6-week-old WT BALB/c mice were divided into six groups (n = 10). Five groups of mice were intraperitoneally injected with 5 plaque forming unit (PFU) of rWT, rT1, rT3, rT4 or r△NSs-eGFP in 100 μL of PBS. As a control, a group of mice were injected with PBS. Mice were monitored for clinical signs and weighed once a day, then humanely euthanized at day 5 or 7. Liver and spleen samples were harvested for analysis. RNA viral loads in liver and spleen were determined by quantitative RT-PCR.
2.10. Quantitative RT-PCR
RNA was extracted from the liver and spleen homogenates using TRIzol Reagent (Life technology, Waltham, MA, USA) according to the manufacturer’s instructions. Quantitative RT-PCR was performed using the HiScript II One Step qRT-PCR SYBR Green Kit (Vazyme, Nanjing, China) and the Bio-Rad CFX96 Real-Time System. Standard curves were drawn using a RVFV RNA positive control obtained through in vitro synthesis.
2.11. Statistical Analyses
Statistical analyses were performed in GraphPad Prism version 6, as defined in the text and figure legends. A p value < 0.05 was considered to be statistically significant.
4. Discussion
Investigating the virulence mechanism of RVFV is critical for understanding its pathogenesis, and NSs was identified to be the key virulence factor in vivo. NSs forms a distinct filament structure in the nuclei of infected cells and it has been proposed that the filament structure formation is associated with its virulence, although the mechanism still awaits further characterization [
22]. Recently, the crystal structure of a truncated NSs protein (83-248 AA) was determined and several mutations on NSs were shown to be detrimental to NSs filament formation (namely T1 (R88D, S228A) and T3 (K150G, T152G)). For this reason, we introduced these mutations into NSs in our system. The NSs T4 mutant (I216D, M219A) was reported to still form nuclear filament and was included in parallel as a control. These mutant viruses showed comparable replication kinetics as theWT virus in vitro. Unexpectedly, we found that the NSs T1 mutant still formed distinct nuclear filament structure in both Vero and Huh7 cells, indicating that this mutation did not affect NSs filament formation in our system. Also different from the previous report, we observed that the NSs T3 mutant displayed a diffusive distribution in the cytoplasm of infected cells, while it was reported to form aggregates in the nuclei of infected cells. The NSs T4 mutant formed nuclear filaments but, unlike the distinct filament structure of WT NSs, the NSs T4 filament appeared as fragmented structures. Currently the reasons for these apparent inconsistencies are unclear; however, it is noteworthy that a virulent wild-type strain was used in this work, whereas the study of Barski and co-workers made use of the attenuated MP-12 strain [
16], which the NSs protein differs from by five amino-acids from the BJ01 strain (
Figure S4). Compared to the other field isolate of RVFV strains, ZH548 and ZH501, the NSs of BJ01 contains four amino-acid substitutions (
Figure S4). Whether these differences in amino acid sequence may lead to different NSs filament formation patterns would be an interesting question for future investigation.
The three NSs mutants showed different intracellular patterns, presenting a unique opportunity to investigate the correlation between NSs filament formation and its virulence. Notably, although the T1 mutant still formed nuclear filaments in infected cells, the virulence of this mutant was strongly reduced in comparison with the WT virus (10% vs. 80%). Similar results were also obtained with the T4 mutant, for which a 30% fatality rate was recorded. On the other hand, the T3 mutant, which was impaired in NSs filament formation, did not cause lethal infection in mice, similar to the r△NSs-eGFP virus. These findings suggest that NSs filament formation is important per se, but probably not sufficient for its in vivo virulence. It remains intriguing how the mutations in T1 and T4 affect virulence. Notably, although replication of the T1 and T4 mutants were comparable with the WT virus in cell culture, replication of these two mutant viruses was significantly reduced in vivo, which may be related to the reduced virulence. Nevertheless, these mutant NSs viruses can be employed for future in-depth investigation of NSs virulence mechanisms.
No nuclear localization signal (NLS) has been identified for NSs so far, and it was proposed that interaction with host factors might mediate nuclear localization [
23]. Interestingly, the NSs T3 mutant showed a cytoplasmic diffusive distribution, indicating that the amino acids K150 and T152 might be critical for mediating NSs nuclear translocation. Comparing the interacting host factors of the WT and T3 NSs might allow for the identification of relevant NSs–host interactions that mediate NSs nuclear translocation. On the one hand, NSs has a molecular weight of 31 kDa, and should be able to freely translocate through the nuclear pore complex [
24]. On the other hand, the T3 mutant was observed nearly entirely in the cytoplasm, indicating that it might form a protein complex with itself and/or with host factors, leading to its sequestration in the cytoplasm. The diffusive distribution of the T3 mutant also indicates that the amino acids K150 and/or T152 might be key amino acids that determine NSs homo-oligomerization and filament formation.
Results from this study also lay the basis for several potential applications. The NSs mutants with compromised virulence might find application in vaccine development to further improve the live attenuated RVFV vaccine candidates. Whether combinations of these NSs mutations would further increase safety and reduce the chance of occurrence of revertant mutations would be interesting questions for future investigation. Additionally, the reporter gene of eGFP was successfully inserted into the reverse genetic system through replacing the NSs coding sequence. This recombinant virus showed robust genetic stability and the reporter gene was efficiently expressed even when the virus had been serially passaged for 15 passages. This recombinant reporter virus has the potential to be used for future anti-RVFV drug screening. These efforts would contribute to the development of effective countermeasures (anti-viral drugs and vaccines) to contain the threat of this highly pathogenic virus from both domesticated animals and humans.


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




