Discovery and Characterization of Novel RNA Viruses in Aquatic North American Wild Birds

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Molecular Methods
2.3. Sequence and Phylogenetic Analyses
2.4. Data Availability
3. Results and Discussion
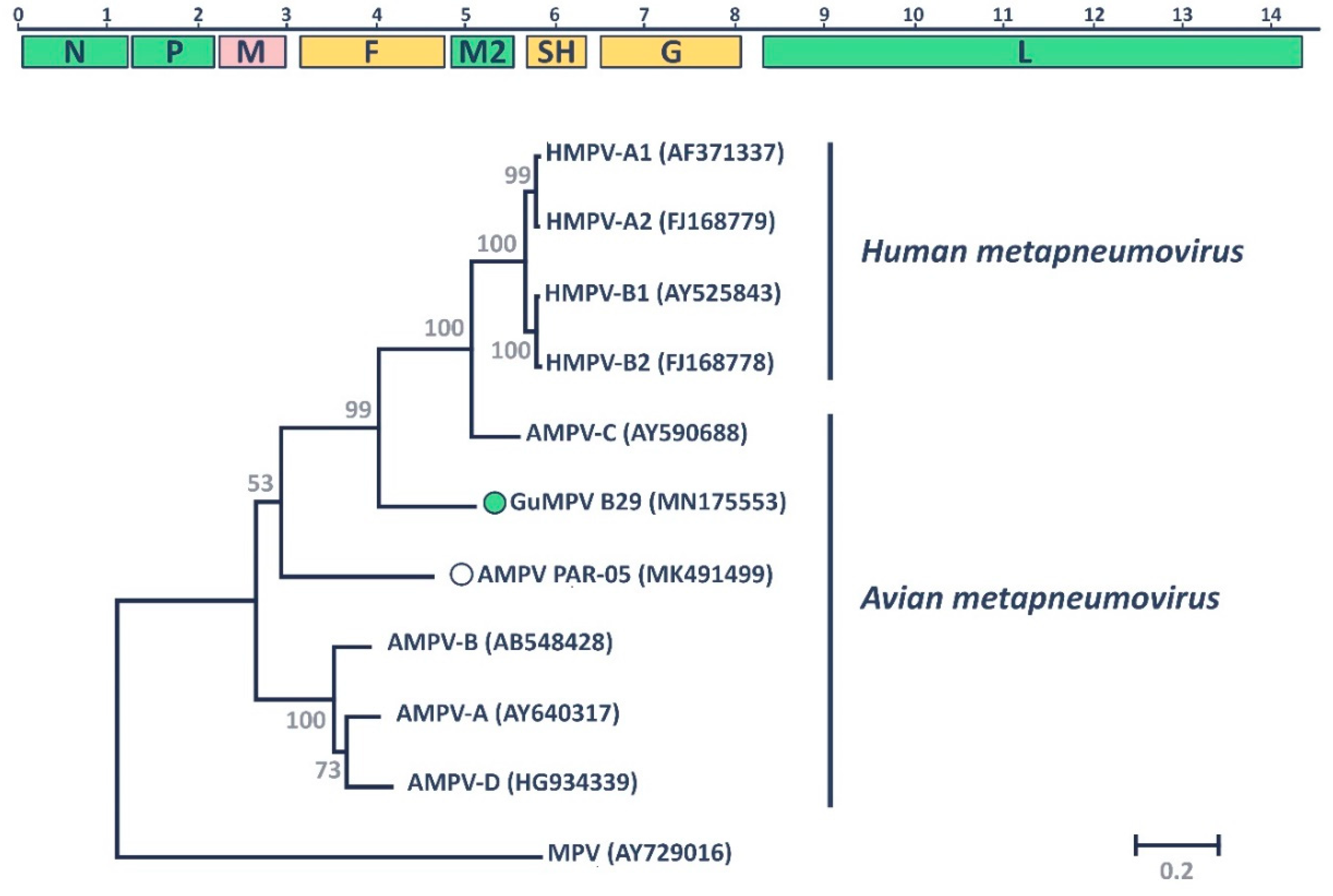
3.1. Gull Metapneumovirus
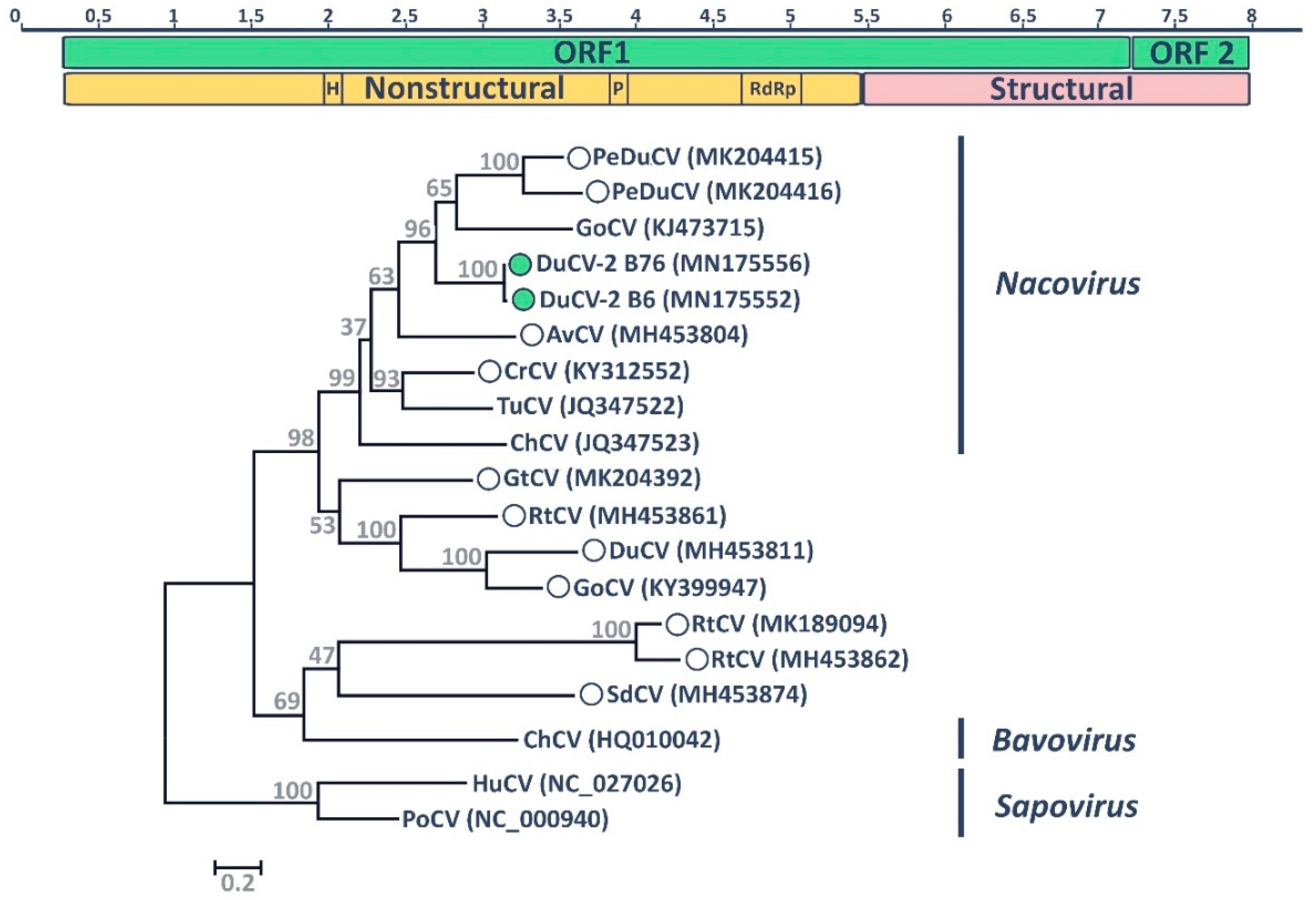
3.2. Gull Coronavirus
3.3. Duck Calicivirus
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prum, R.O.; Berv, J.S.; Dornburg, A.; Field, D.J.; Townsend, J.P.; Lemmon, E.M.; Lemmon, A.R. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 2015, 526, 569–573. [Google Scholar] [CrossRef]
- Şekercioğlu, Ç.H.; Daily, G.C.; Ehrlich, P.R. Ecosystem consequences of bird declines. Proc. Natl. Acad. Sci. USA 2004, 101, 18042–18047. [Google Scholar] [CrossRef] [PubMed]
- Viana, D.S.; Santamaría, L.; Figuerola, J. Migratory birds as global dispersal vectors. Trends Ecol. Evol. 2016, 31, 763–775. [Google Scholar] [CrossRef]
- Bauer, S.; Hoye, B.J. Migratory animals couple biodiversity and ecosystem functioning worldwide. Science 2014, 344, 1242552. [Google Scholar] [CrossRef] [PubMed]
- Canuti, M.; Munro, H.J.; Robertson, G.J.; Kroyer, A.N.K.; Roul, S.; Ojkic, D.; Whitney, H.G.; Lang, A.S. New insight into avian papillomavirus ecology and evolution from characterization of novel wild bird papillomaviruses. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.M.; Ferree, E.; Simon, D.M.; Yeh, P.J. Patterns of bird-bacteria associations. Ecohealth 2018, 15, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.; Munster, V.J.; Wallensten, A.; Waldenström, J.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Global patterns of influenza A virus in wild birds. Science 2006, 312, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Reed, K.D.; Meece, J.K.; Henkel, J.S.; Shukla, S.K. Birds, migration and emerging zoonoses: West Nile virus, Lyme disease, influenza A and enteropathogens. Clin. Med. Res. 2003, 1, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Chamings, A.; Nelson, T.M.; Vibin, J.; Wille, M.; Klaassen, M.; Alexandersen, S. Detection and characterisation of coronaviruses in migratory and non-migratory Australian wild birds. Sci. Rep. 2018, 8, 5980. [Google Scholar] [CrossRef] [PubMed]
- Bodewes, R. Novel viruses in birds: Flying through the roof or is a cage needed? Veterinary J. 2018, 233, 55–62. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; To, K.K.-W.; Chen, H.; Yuen, K.-Y. Cross-species transmission and emergence of novel viruses from birds. Curr. Opin. Virol. 2015, 10, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Wille, M.; Eden, J.-S.; Shi, M.; Klaassen, M.; Hurt, A.C.; Holmes, E.C. Virus–virus interactions and host ecology are associated with RNA virome structure in wild birds. Mol. Ecol. 2018, 27, 5263–5278. [Google Scholar] [CrossRef]
- Verhoeven, J.T.P.; Canuti, M.; Munro, H.J.; Dufour, S.C.; Lang, A.S. ViDiT-CACTUS: An inexpensive and versatile library preparation and sequence analysis method for virus discovery and other microbiology applications. Can. J. Microbiol. 2018, 64, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Rima, B.; Collins, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.A.; Lee, B.; Maisner, A.; Rota, P.; Wang, L.; et al. ICTV Virus Taxonomy Profile: Pneumoviridae. J. Gen. Virol. 2017, 98, 2912–2913. [Google Scholar] [CrossRef] [PubMed]
- Retallack, H.; Clubb, S.; DeRisi, J.L. Genome sequence of a divergent avian metapneumovirus from a monk parakeet (Myiopsitta monachus). Microbiol. Resour. Announc. 2019, 8, e00284-19. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.K. Avian pneumovirus infections of turkeys and chickens. Vet. J. 2000, 160, 118–125. [Google Scholar] [CrossRef]
- Umar, S.; Sabir, H.; Ahmed, A.; Subhan, S. Avian metapneumovirus infection in poultry. World’s Poultry Sci. J. 2016, 72, 833–846. [Google Scholar] [CrossRef]
- Jardine, C.M.; Parmley, E.J.; Buchanan, T.; Nituch, L.; Ojkic, D. Avian metapneumovirus subtype C in wild waterfowl in Ontario, Canada. Transbound. Emerg. Dis. 2018, 65, 1098–1102. [Google Scholar] [CrossRef]
- Bennett, R.S.; Nezworski, J.; Velayudhan, B.T.; Nagaraja, K.V.; Zeman, D.H.; Dyer, N.; Graham, T.; Lauer, D.C.; Njenga, M.K.; Halvorson, D.A. Evidence of avian pneumovirus spread beyond Minnesota among wild and domestic birds in central North America. Avian Dis. 2004, 48, 902–908. [Google Scholar] [CrossRef]
- van Boheemen, S.; Bestebroer, T.M.; Verhagen, J.H.; Osterhaus, A.D.M.E.; Pas, S.D.; Herfst, S.; Fouchier, R.A.M. A family-wide RT-PCR assay for detection of paramyxoviruses and application to a large-scale surveillance study. PLoS ONE 2012, 7, e34961. [Google Scholar] [CrossRef]
- Rizotto, L.S.; Simão, R.M.; Scagion, G.P.; Simasaki, A.A.; Caserta, L.C.; Benassi, J.C.; Arns, C.W.; Ferreira, H.L.; Rizotto, L.S.; Simão, R.M.; et al. Detection of avian metapneumovirus subtype A from wild birds in the State of São Paulo, Brazil. Pesquisa Veterinária Brasileira 2019, 39, 209–213. [Google Scholar] [CrossRef]
- Shin, H.-J.; Njenga, M.K.; McComb, B.; Halvorson, D.A.; Nagaraja, K.V. Avian pneumovirus (APV) RNA from wild and sentinel birds in the United States has genetic homology with RNA from APV isolates from domestic turkeys. J. Clin. Microbiol. 2000, 38, 4282–4284. [Google Scholar]
- ICTV Coronaviridae, Virus Taxonomy: 2018b Release. Available online: https://talk.ictvonline.org/ (accessed on 17 May 2019).
- Miłek, J.; Blicharz-Domańska, K. Coronaviruses in avian species – review with focus on epidemiology and diagnosis in wild birds. J. Vet. Res. 2018, 62, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.K.A.; Jackwood, M.; Jones, R.C. The long view: 40 years of infectious bronchitis research. Avian Pathol. 2012, 41, 239–250. [Google Scholar] [CrossRef]
- Chen, Y.-N.; Wu, C.C.; Lin, T.L. Turkey coronavirus: An updated review. Taiwan Vet. J. 2015, 41, 1–10. [Google Scholar] [CrossRef]
- Abdelmohsen Rohaim, M.; Fawzi El Naggar, R.; Maher Helal, A.; Mohamed Bayoumi, M.; Ahmed El-Saied, M.; Abdelazez Ahmed, K.; Zubair Shabbir, M.; Munir, M. Genetic diversity and phylodynamics of avian coronaviruses in Egyptian wild birds. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Leung, C.Y.H.; Gilbert, M.; Joyner, P.H.; Ng, E.M.; Tse, T.M.; Guan, Y.; Peiris, J.S.M.; Poon, L.L.M. Avian coronavirus in wild aquatic birds. J. Virol. 2011, 85, 12815–12820. [Google Scholar] [CrossRef]
- de Sales Lima, F.E.; Gil, P.; Pedrono, M.; Minet, C.; Kwiatek, O.; Campos, F.S.; Spilki, F.R.; Roehe, P.M.; Franco, A.C.; Maminiaina, O.F.; et al. Diverse gammacoronaviruses detected in wild birds from Madagascar. Eur. J. Wildl. Res. 2015, 61, 635–639. [Google Scholar] [CrossRef]
- Hughes, L.A.; Savage, C.; Naylor, C.; Bennett, M.; Chantrey, J.; Jones, R. Genetically diverse coronaviruses in wild bird populations of Northern England. Emerg. Infect. Dis. 2009, 15, 1091–1094. [Google Scholar] [CrossRef]
- Muradrasoli, S.; Mohamed, N.; Hornyák, Á.; Fohlman, J.; Olsen, B.; Belák, S.; Blomberg, J. Broadly targeted multiprobe QPCR for detection of coronaviruses: Coronavirus is common among mallard ducks (Anas platyrhynchos). J. Virol. Methods 2009, 159, 277–287. [Google Scholar] [CrossRef]
- Chen, G.-Q.; Zhuang, Q.-Y.; Wang, K.-C.; Liu, S.; Shao, J.-Z.; Jiang, W.-M.; Hou, G.-Y.; Li, J.-P.; Yu, J.-M.; Li, Y.-P.; et al. Identification and survey of a novel avian coronavirus in ducks. PLoS ONE 2013, 8, e72918. [Google Scholar] [CrossRef]
- Papineau, A.; Berhane, Y.; Wylie, T.N.; Wylie, K.M.; Sharpe, S.; Lung, O. Genome organization of Canada goose coronavirus, a novel species identified in a mass die-off of Canada geese. Sci. Rep. 2019, 9, 5954. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Q.-Y.; Wang, K.-C.; Liu, S.; Hou, G.-Y.; Jiang, W.-M.; Wang, S.-C.; Li, J.-P.; Yu, J.-M.; Chen, J.-M. Genomic analysis and surveillance of the coronavirus dominant in ducks in China. PLoS ONE 2015, 10, e0129256. [Google Scholar] [CrossRef]
- ICTV Caliciviridae, Virus Taxonomy: 2018b release. Available online: https://talk.ictvonline.org/ (accessed on 17 May 2019).
- Day, J.M.; Ballard, L.L.; Duke, M.V.; Scheffler, B.E.; Zsak, L. Metagenomic analysis of the turkey gut RNA virus community. Virol. J. 2010, 7, 313. [Google Scholar] [CrossRef]
- Desselberger, U. Caliciviridae other than noroviruses. Viruses 2019, 11, 286. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.; Reetz, J.; Otto, P. Genetic characterization of a novel calicivirus from a chicken. Arch. Virol. 2011, 156, 1143–1150. [Google Scholar] [CrossRef]
- Wolf, S.; Reetz, J.; Hoffmann, K.; Gründel, A.; Schwarz, B.-A.; Hänel, I.; Otto, P.H. Discovery and genetic characterization of novel caliciviruses in German and Dutch poultry. Arch. Virol. 2012, 157, 1499–1507. [Google Scholar] [CrossRef]
- Liao, Q.; Wang, X.; Wang, D.; Zhang, D. Complete genome sequence of a novel calicivirus from a goose. Arch. Virol. 2014, 159, 2529–2531. [Google Scholar] [CrossRef]
- de Souza, W.M.; Fumagalli, M.J.; de Araujo, J.; Ometto, T.; Modha, S.; Thomazelli, L.M.; Durigon, E.L.; Murcia, P.R.; Figueiredo, L.T.M. Discovery of novel astrovirus and calicivirus identified in ruddy turnstones in Brazil. Sci. Rep. 2019, 9, 5556. [Google Scholar] [CrossRef]
- Wille, M.; Shi, M.; Klaassen, M.; Hurt, A.; Holmes, E. Virome heterogeneity and connectivity in waterfowl and shorebird communities. ISME J. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, R.; Serra, F.; Tárraga, J.; Medina, I.; Carbonell, J.; Pulido, L.; de María, A.; Capella-Gutíerrez, S.; Huerta-Cepas, J.; Gabaldón, T.; et al. Phylemon 2.0: A suite of web-tools for molecular evolution, phylogenetics, phylogenomics and hypotheses testing. Nucleic Acids Res. 2011, 39, W470–W474. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Yun, B.; Guan, X.; Liu, Y.; Gao, Y.; Wang, Y.; Qi, X.; Cui, H.; Liu, C.; Zhang, Y.; Gao, L.; et al. Trypsin- and low pH-mediated fusogenicity of avian metapneumovirus fusion proteins is determined by residues at positions 100, 101 and 294. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- van den Hoogen, B.G.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Analysis of the genomic sequence of a human metapneumovirus. Virology 2002, 295, 119–132. [Google Scholar] [CrossRef]
- Turpin, E.A.; Stallknecht, D.E.; Slemons, R.D.; Zsak, L.; Swayne, D.E. Evidence of avian metapneumovirus subtype C infection of wild birds in Georgia, South Carolina, Arkansas and Ohio, USA. Avian Pathol. 2008, 37, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Heffels-Redmann, U.; Neumann, U.; Braune, S.; Cook, J.K.A.; Prüter, J. Serological evidence for susceptibility of sea gulls to avian pneumovirus (APV) infection. Proceedings international symposium on infectious bronchitis and pneumovirus infections in poultry, Rauischholzhausen, Germany, 15–18 June 1998; pp. 23–25. [Google Scholar]
- Fang, S.; Shen, H.; Wang, J.; Tay, F.P.L.; Liu, D.X. Functional and genetic studies of the substrate specificity of coronavirus infectious bronchitis virus 3C-like proteinase. J. Virol. 2010, 84, 7325–7336. [Google Scholar] [CrossRef]
- Gadlage, M.J.; Denison, M.R. Exchange of the coronavirus replicase polyprotein cleavage sites alters protease specificity and processing. J. Virol. 2010, 84, 6894–6898. [Google Scholar] [CrossRef] [PubMed]
- Kindler, E.; Thiel, V. To sense or not to sense viral RNA--essentials of coronavirus innate immune evasion. Curr. Opin. Microbiol. 2014, 20, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.-C.; Lin, M.-H.; Chuang, C.-Y.; Hsu, C.-H. Macro domain from Middle East Respiratory Syndrome Coronavirus (MERS-CoV) is an efficient ADP-ribose binding module: Crystal structure and biochemical studies. J. Biol. Chem. 2016, 291, 4894–4902. [Google Scholar] [CrossRef]
- Keep, S.; Bickerton, E.; Armesto, M.; Britton, P. The ADRP domain from a virulent strain of infectious bronchitis virus is not sufficient to confer a pathogenic phenotype to the attenuated Beaudette strain. J. Gen. Virol. 2018, 99, 1097–1102. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Snijder, E.J.; Gorbalenya, A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef] [PubMed]
- Domanska-Blicharz, K.; Jacukowicz, A.; Lisowska, A.; Wyrostek, K.; Minta, Z. Detection and molecular characterization of infectious bronchitis-like viruses in wild bird populations. Avian Pathol. 2014, 43, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Hepojoki, S.; Lindh, E.; Vapalahti, O.; Huovilainen, A. Prevalence and genetic diversity of coronaviruses in wild birds, Finland. Infect. Ecol. Epidemiol. 2017, 7, 1408360. [Google Scholar] [CrossRef] [PubMed]
- Wille, M.; Muradrasoli, S.; Nilsson, A.; Järhult, J.D. High prevalence and putative lineage maintenance of avian coronaviruses in Scandinavian waterfowl. PLoS ONE 2016, 11, e0150198. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.J.; Hilt, D.A.; Poulson, R.; Stallknecht, D.E.; Jackwood, M.W. Identification of avian coronavirus in wild aquatic birds of the central and eastern USA. JWDI 2015, 51, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-R.; Oem, J.-K. Surveillance of avian coronaviruses in wild bird populations of Korea. J. Wildl. Dis. 2014, 50, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Canuti, M.; Lang, A.S.; Memorial University of Newfoundland, 232 Elizabeth Ave., St. John’s, NL A1B 3X9, Canada. unpublished work. 2019.
- L’Homme, Y.; Sansregret, R.; Plante-Fortier, E.; Lamontagne, A.-M.; Ouardani, M.; Lacroix, G.; Simard, C. Genomic characterization of swine caliciviruses representing a new genus of Caliciviridae. Virus Genes 2009, 39, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Mikalsen, A.B.; Nilsen, P.; Frøystad-Saugen, M.; Lindmo, K.; Eliassen, T.M.; Rode, M.; Evensen, Ø. Characterization of a novel calicivirus causing systemic infection in Atlantic salmon (Salmo salar L.): Proposal for a new genus of Caliciviridae. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, M.; Dong, Y.; Zhang, B.; Zhang, D. Genetic characterization of a novel calicivirus from a goose. Arch. Virol. 2017, 162, 2115–2118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, S.; Liu, D.; Zhou, C.; Li, W.; Lin, Y.; Wang, X.; Shen, Q.; Wang, H.; Li, C.; et al. The fecal virome of red-crowned cranes. Arch. Virol. 2019, 164, 3–16. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5’–3’) | nt Position | Reference Genome |
|---|---|---|---|
| Gull metapneumovirus | |||
| Meta_F15 | TCAGATGGGTCTTCAAAGGTG | 11957–11977 | MN175553 |
| Meta_F16 | GCATAGACTGTCTGTCAGTAG | 12084–12104 | |
| Meta_R20 | TACTCGTTGCACTGACTCCG | 12246–12265 | |
| Gull coronavirus | |||
| Corona_F4 | CTGTTGAGGTTGACGAACAAGG | 1828–1849 | MN175554 |
| Corona_R5 | AGTAACAGTCTTACCACCAGC | 2040–2060 | |
| Corona_ R4 | GCACCAACTTGCGACATTGG | 2001–2020 | |
| Duck calicivirus | |||
| Calici_F8 | GATCTGGCATGTATGGAGGC | 5851–5870 | MN175552 |
| Calici_F7 | TCCTTCCACCAGGCATCAAC | 5887–5906 | |
| Calici_R13 | GGTAGTGGTTCCAGGAGTAG | 6284–6303 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canuti, M.; Kroyer, A.N.K.; Ojkic, D.; Whitney, H.G.; Robertson, G.J.; Lang, A.S. Discovery and Characterization of Novel RNA Viruses in Aquatic North American Wild Birds. Viruses 2019, 11, 768. https://doi.org/10.3390/v11090768
Canuti M, Kroyer ANK, Ojkic D, Whitney HG, Robertson GJ, Lang AS. Discovery and Characterization of Novel RNA Viruses in Aquatic North American Wild Birds. Viruses. 2019; 11(9):768. https://doi.org/10.3390/v11090768
Chicago/Turabian StyleCanuti, Marta, Ashley N. K. Kroyer, Davor Ojkic, Hugh G. Whitney, Gregory J. Robertson, and Andrew S. Lang. 2019. "Discovery and Characterization of Novel RNA Viruses in Aquatic North American Wild Birds" Viruses 11, no. 9: 768. https://doi.org/10.3390/v11090768
APA StyleCanuti, M., Kroyer, A. N. K., Ojkic, D., Whitney, H. G., Robertson, G. J., & Lang, A. S. (2019). Discovery and Characterization of Novel RNA Viruses in Aquatic North American Wild Birds. Viruses, 11(9), 768. https://doi.org/10.3390/v11090768