Effects of the Q80K Polymorphism on the Physicochemical Properties of Hepatitis C Virus Subtype 1a NS3 Protease

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Analysis and Statistical Treatment
2.2. Comparative Modeling
2.3. Molecular Dynamics (MD) Simulations
2.4. Trajectory Analysis
2.5. Correlation Network Analysis
2.6. Phylogenetic Analysis
3. Results
3.1. Sequence Analysis, Statistical Treatment, and Comparative Modeling
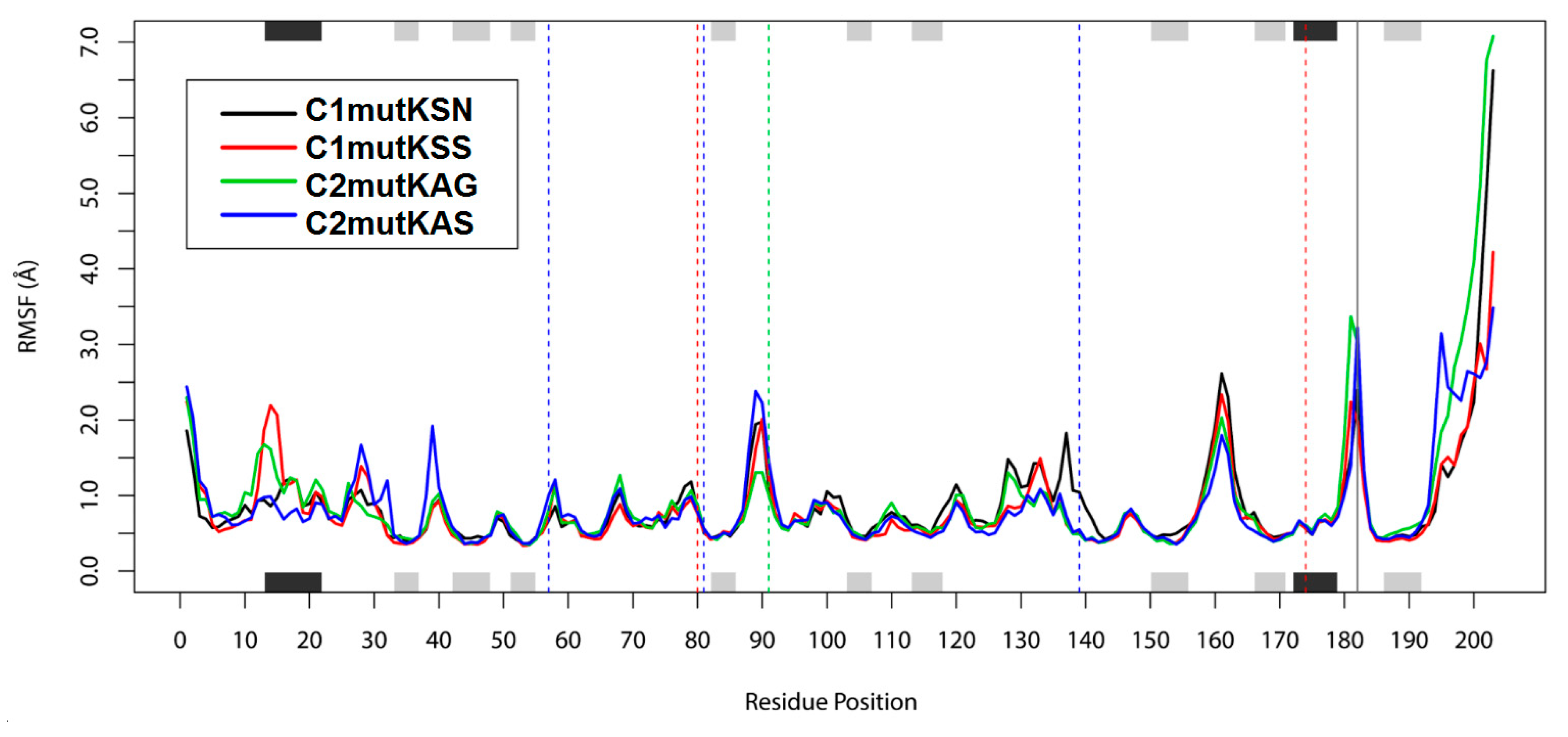
3.2. Assessing Dynamic Properties of the Models in Aqueous Solution
3.3. Hydrogen Bonding Interactions
3.4. Correlation Network Analysis and Community Network Comparison
3.5. Phylogenetic Analysis
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karnsakul, W.; Schwarz, K.B. Hepatitis B and C. Pediatr. Clin. N. Am. 2017, 64, 641–658. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Agbowuro, A.A.; Huston, W.M.; Gamble, A.B.; Tyndall, J.D.A. Proteases and protease inhibitors in infectious diseases. Med. Res. Rev. 2018, 38, 1295–1331. [Google Scholar] [CrossRef] [PubMed]
- Sorbo, M.C.; Cento, V.; Di Maio, V.C.; Howe, A.Y.M.; Garcia, F.; Perno, C.F.; Ceccherini-Silberstein, F. Hepatitis C virus drug resistance associated substitutions and their clinical relevance: Update 2018. Drug Resist. Updates 2018, 40, 40–41. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J. Hepatol. 2016, 64, 486–504. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M. Hepatitis C virus resistance to direct-acting antiviral drugs in interferon-free regimens. Gastroenterology 2016, 151, 70–86. [Google Scholar] [CrossRef]
- Suzuki, T.; Aizaki, H.; Murakami, K.; Shoji, I.; Wakita, T. Molecular biology of hepatitis C virus. J. Gastroenterol. 2007, 42, 411–423. [Google Scholar] [CrossRef]
- Cento, V.; Chevaliez, S.; Perno, C.F. Resistance to direct-acting antiviral agents: Clinical utility and significance. Curr. Opin. HIV AIDS 2015, 10, 381–389. [Google Scholar] [CrossRef]
- Pham, L.V.; Jensen, S.B.; Fahnøe, U.; Pedersen, M.S.; Tang, Q.; Ghanem, L.; Ramirez, S.; Humes, D.; Serre, S.B.N.; Schønning, K.; et al. HCV genotype 1-6 NS3 residue 80 substitutions impact protease inhibitor activity and promote viral escape. J. Hepatol. 2019, 70, 388–397. [Google Scholar] [CrossRef]
- Lenz, O.; Verbinnen, T.; Fevery, B.; Tambuyzer, L.; Vijgen, L.; Peeters, M.; Buelens, A.; Ceulemans, H.; Beumont, M.; Picchio, G.; et al. Virology analyses of HCV isolates from genotype 1-infected patients treated with simeprevir plus peginterferon/ribavirin in Phase IIb/III studies. J. Hepatol. 2015, 62, 1008–1014. [Google Scholar] [CrossRef]
- Pickett, B.E.; Striker, R.; Lefkowitz, E.J. Evidence for separation of HCV subtype 1a into two distinct clades. J. Viral Hepat. 2011, 18, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Peres-da-Silva, A.; Almeida, A.J.; Lampe, E. Genetic diversity of NS3 protease from Brazilian HCV isolates and possible implications for therapy with direct-acting antiviral drugs. Mem. Inst. Oswaldo Cruz 2012, 107, 254–261. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Luca, A.; Di Giambenedetto, S.; Lo Presti, A.; Sierra, S.; Prosperi, M.; Cella, E.; Giovanetti, M.; Torti, C.; Caudai, C.; Vicenti, I.; et al. Two distinct hepatitis c virus genotype 1a clades have different geographical distribution and association with natural resistance to NS3 protease inhibitors. Open Forum Infect. Dis. 2015, 2, ofv043. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.F.; Bello, G.; Vidal, L.L.; Souza, S.L.; Mir, D.; Soares, M.A. In-depth phylogenetic analysis of hepatitis C virus subtype 1a and occurrence of 80K and associated polymorphisms in the NS3 protease. Sci. Rep. 2016, 6, 31780. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, P.; Moretti, S.; Xenarios, I.; Orobitg, M.; Montanyola, A.; Chang, J.M.; Taly, J.F.; Notredame, C. T-Coffee: A web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011, 39, W13–W17. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Myers, G. Signature pattern analysis: A method for assessing viral sequence relatedness. AIDS Res. Hum. Retrovir. 1992, 8, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Bogen, S.L.; Ruan, S.; Liu, R.; Agrawal, S.; Pichardo, J.; Prongay, A.; Baroudy, B.; Saksena, A.K.; Girijavallabhan, V.; Njoroge, F.G. Depeptidization efforts on P3-P2’ alpha-ketoamide inhibitors of HCV NS3-4A serine protease: Effect on HCV replicon activity. Bioorg. Med. Chem. Lett. 2006, 16, 1621–1627. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2017, 1654, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Benkert, P.; Künzli, M.; Schwede, T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009, 37, W510–W514. [Google Scholar] [CrossRef] [PubMed]
- Bond, C.S.; Schüttelkopf, A.W. ALINE: A WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2005, 1–2, 19–25. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing. 2017. Available online: http://www.R-project.org/ (accessed on 30 October 2018).
- Csárdi, G.; Nepusz, T. The igraph software package for complex network research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Girvan, M.; Newman, M.E. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA 2002, 99, 7821–7826. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef]
- Martínez, A.P.; Culasso, A.C.A.; Pérez, P.S.; Romano, V.; Campos, R.H.; Ridruejo, E.; García, G.; Di Lello, F.A. Polymorphisms associated with resistance to protease inhibitors in naïve patients infected with hepatitis C virus genotype 1 in Argentina: Low prevalence of Q80K. Virus Res. 2017, 240, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA: Protein complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, R.M.; Liang, R.H.; Joy, J.B.; Krajden, M.; Montaner, J.S.; Harrigan, P.R.; Poon, A.F. Global origin and transmission of hepatitis C virus nonstructural protein 3 Q80K polymorphism. J. Infect. Dis. 2015, 211, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Welsch, C. Genetic barrier and variant fitness in hepatitis C as critical parameters for drug resistance development. Drug Discov. Today Technol. 2014, 11, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Murai, K.; Shimakami, T.; Welsch, C.; Shirasaki, T.; Liu, F.; Kitabayashi, J.; Tanaka, S.; Funaki, M.; Omura, H.; Nishikawa, T.; et al. Unexpected replication boost by simeprevir for simeprevir-resistant variants in genotype 1a hepatitis C virus. Antimicrob. Agents Chemother. 2018, 62, e02601-17. [Google Scholar] [CrossRef]
- Sun, J.H.; O’Boyle Ii, D.R.; Zhang, Y.; Wang, C.; Nower, P.; Valera, L.; Roberts, S.; Nettles, R.E.; Fridell, R.A.; Gao, M. Impact of a baseline polymorphism on the emergence of resistance to the hepatitis C virus nonstructural protein 5A replication complex inhibitor, BMS-790052. Hepatology 2012, 55, 1692–1699. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | DOPE Score | ERRAT2 | QMean | Ramachandran Plot | ||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | ||||
| 2f9v | −22100 | 94.047 | −0.35 | 95.5 | 4.5 | 0.0 |
| c1mutKSN | −21543 | 83.132 | 0.19 | 97.9 | 2.1 | 0.0 |
| c1mutKSS | −21453 | 92.261 | 0.21 | 98.5 | 1.5 | 0.0 |
| c2mutKAG | −21581 | 91.071 | 0.13 | 98.5 | 1.5 | 0.0 |
| c2mutKAS | −21665 | 89.881 | 0.44 | 98.5 | 1.5 | 0.0 |
| Atoms | Occupancy (%) | ||||
|---|---|---|---|---|---|
| Donor | Acceptor | c1mutKSN | c1mutKSS | c2mutKAG | c2mutKAS |
| HIS57–ND1 | ASP81–CG | - | 10.34 | - | 48.99 |
| HIS57–ND1 | ASP81–OD1 | - | 6.38 | - | 27.41 |
| HIS57–ND1 | ASP81–OD2 | - | 7.64 | - | 20.04 |
| SER139–OG | LYS136–O | 13.14 | 26.55 | 6.58 | 20.44 |
| Atoms | Distance (Å) | ||||
|---|---|---|---|---|---|
| Donor | Acceptor | c1mutKSN | c1mutKSS | c2mutKAG | c2mutKAS |
| HIS57–ND1 | ASP81–CG | 4.97 ± 1.00 | 5.69 ± 1.75 | 5.41 ± 1.34 | 4.95 ± 1.89 |
| HIS57–ND1 | ASP81–OD1 | 5.13 ± 0.96 | 5.87 ± 1.89 | 5.76 ± 1.36 | 4.87 ± 2.14 |
| HIS57–ND1 | ASP81–OD2 | 5.27 ± 1.00 | 5.83 ± 1.94 | 5.73 ± 1.37 | 5.02 ± 2.10 |
| SER139–OG | LYS136–O | 4.35 ± 1.81 | 4.33 ± 1.97 | 5.20 ± 1.99 | 4.88 ± 2.12 |
| c1mutKSN | c1mutKSS | c2mutKAG | c2mutKAS | |
|---|---|---|---|---|
| c1mutKSN | 1 | 0.62 | 0.37 | 0.42 |
| c1mutKSS | 1 | 0.57 | 0.47 | |
| c2mutKAG | 1 | 0.46 | ||
| c2mutKAS | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peres-da-Silva, A.; Antunes, D.; Quintanilha Torres, A.L.; Caffarena, E.R.; Lampe, E. Effects of the Q80K Polymorphism on the Physicochemical Properties of Hepatitis C Virus Subtype 1a NS3 Protease. Viruses 2019, 11, 691. https://doi.org/10.3390/v11080691
Peres-da-Silva A, Antunes D, Quintanilha Torres AL, Caffarena ER, Lampe E. Effects of the Q80K Polymorphism on the Physicochemical Properties of Hepatitis C Virus Subtype 1a NS3 Protease. Viruses. 2019; 11(8):691. https://doi.org/10.3390/v11080691
Chicago/Turabian StylePeres-da-Silva, Allan, Deborah Antunes, André Luiz Quintanilha Torres, Ernesto Raul Caffarena, and Elisabeth Lampe. 2019. "Effects of the Q80K Polymorphism on the Physicochemical Properties of Hepatitis C Virus Subtype 1a NS3 Protease" Viruses 11, no. 8: 691. https://doi.org/10.3390/v11080691
APA StylePeres-da-Silva, A., Antunes, D., Quintanilha Torres, A. L., Caffarena, E. R., & Lampe, E. (2019). Effects of the Q80K Polymorphism on the Physicochemical Properties of Hepatitis C Virus Subtype 1a NS3 Protease. Viruses, 11(8), 691. https://doi.org/10.3390/v11080691