Viral Long-Term Evolutionary Strategies Favor Stability over Proliferation


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Retrieval
2.2. Phylogenetic Analyses
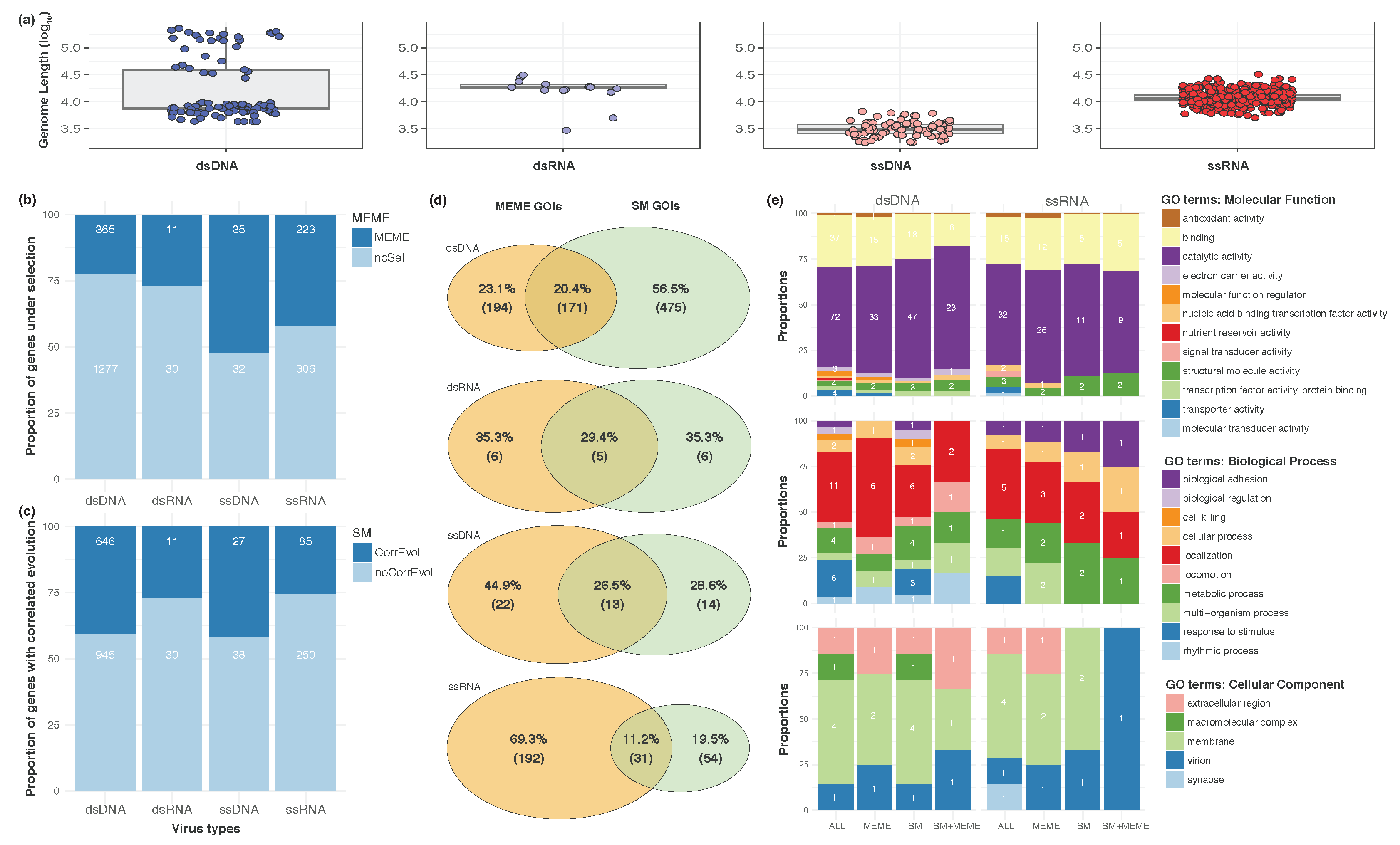
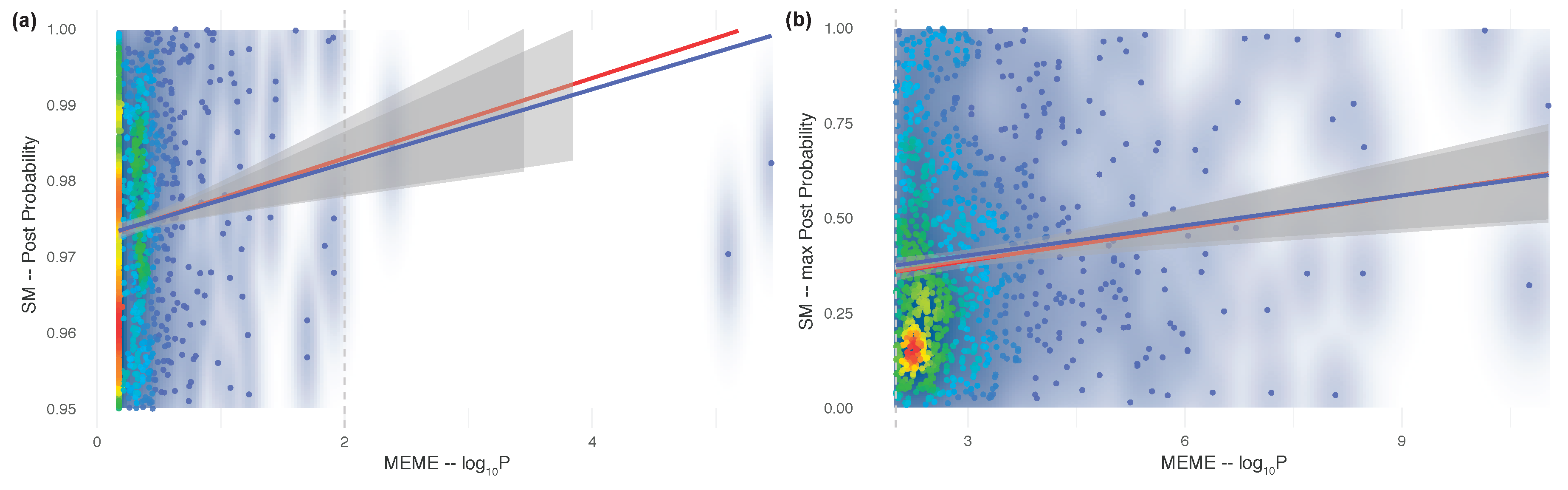
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BGM | Bayesian graphical model |
| GO | Gene ontology |
| dsDNA | Double stranded DNA |
| dsRNA | Double stranded RNA |
| MEME | Mixed effects model of evolution (analysis of positive selection) |
| SEM | Standard error of the mean |
| SM | SpiderMonkey (analysis of correlated evolution) |
| ssDNA | Single stranded DNA |
| ssRNA | Single stranded RNA |
References
- Smith, G.J.D.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Gire, S.K.; Goba, A.; Andersen, K.G.; Sealfon, R.S.G.; Park, D.J.; Kanneh, L.; Jalloh, S.; Momoh, M.; Fullah, M.; Dudas, G.; et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014, 345, 1369–1372. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Azevedo, R.D.S.D.S.; Kraemer, M.U.G.; Souza, R.; Cunha, M.S.; Hill, S.C.; Thézé, J.; Bonsall, M.B.; Bowden, T.A.; Rissanen, I.; et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science 2016, 352, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Sabino, E.C.; Nunes, M.R.T.; Alcantara, L.C.J.; Loman, N.J.; Pybus, O.G. Mobile real-time surveillance of Zika virus in Brazil. Genome Med. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Grenfell, B.T.; Pybus, O.G.; Gog, J.R.; Wood, J.L.N.; Daly, J.M.; Mumford, J.A.; Holmes, E.C. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 2004, 303, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C. The Evolution and Emergence of RNA Viruses; Oxford Series in Ecology and Evolution; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Holmes, E.C. What does virus evolution tell us about virus origins? J. Virol. 2011, 85, 5247–5251. [Google Scholar] [CrossRef] [PubMed]
- Kryazhimskiy, S.; Dushoff, J.; Bazykin, G.A.; Plotkin, J.B. Prevalence of epistasis in the evolution of influenza A surface proteins. PLoS Genet. 2011, 7, e1001301. [Google Scholar] [CrossRef] [PubMed]
- Maddison, W.P.; FitzJohn, R.G. The unsolved challenge to phylogenetic correlation tests for categorical characters. Syst. Biol. 2015, 64, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.I.; Suchard, M.A.; Bloom, J.D. Stability-mediated epistasis constrains the evolution of an influenza protein. Elife 2013, 2, e00631. [Google Scholar] [CrossRef]
- Meer, M.V.; Kondrashov, A.S.; Artzy-Randrup, Y.; Kondrashov, F.A. Compensatory evolution in mitochondrial tRNAs navigates valleys of low fitness. Nature 2010, 464, 279–282. [Google Scholar] [CrossRef]
- Dench, J.; Hinz, A.; Aris-Brosou, S.; Kassen, R. The idiosyncratic drivers of correlated evolution. bioRxiv 2019, 2019, 474536. [Google Scholar]
- Li, C.; Qian, W.; Maclean, C.J.; Zhang, J. The fitness landscape of a tRNA gene. Science 2016, 352, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Nshogozabahizi, J.C.; Dench, J.; Aris-Brosou, S. Widespread historical contingency in Influenza viruses. Genetics 2017, 205, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Lyons, D.M.; Lauring, A.S. Mutation and epistasis in Influenza virus evolution. Viruses 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Aris-Brosou, S.; Ibeh, N.; Noël, J. Viral outbreaks involve destabilized evolutionary networks: Evidence from Ebola, Influenza and Zika. Sci. Rep. 2017, 7, 11881. [Google Scholar] [CrossRef] [PubMed]
- Ibeh, N.; Nshogozabahizi, J.C.; Aris-Brosou, S. Both epistasis and diversifying selection drive the structural evolution of the Ebola virus glycoprotein mucin-like domain. J. Virol. 2016, 90, 5475–5484. [Google Scholar] [CrossRef] [PubMed]
- Stano, M.; Beke, G.; Klucar, L. viruSITE-integrated database for viral genomics. Database 2016, 2016. [Google Scholar] [CrossRef]
- Aris-Brosou, S. Available online: https://github.com/sarisbro (accessed on 30 May 2019).
- NCBI. Available online: https://ftp.ncbi.nih.gov/genbank/ (accessed on 30 May 2019).
- Gilbert, D. Sequence file format conversion with command-line readseq. Curr. Protoc. Bioinform. 2003. [Google Scholar] [CrossRef]
- van Boheemen, S.; de Graaf, M.; Lauber, C.; Bestebroer, T.M.; Raj, V.S.; Zaki, A.M.; Osterhaus, A.D.M.E.; Haagmans, B.L.; Gorbalenya, A.E.; Snijder, E.J.; et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. MBio 2012, 3. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Staton, E. Available online: https://github.com/sestaton/HMMER2GO (accessed on 30 May 2019).
- Pfam. Available online: http://ftp.ebi.ac.uk/pub/databases/Pfam/current_release/Pfam-A.hmm.gz (accessed on 30 May 2019).
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenführer, J. Gene set enrichment analysis with topGO. Bioconduct. Improv. 2009, 27, 1–26. [Google Scholar]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Aris-Brosou, S.; Rodrigue, N. The essentials of computational molecular evolution. Methods Mol. Biol. 2012, 855, 111–152. [Google Scholar] [CrossRef]
- Revell, L.J. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Poon, A.F.Y.; Lewis, F.I.; Frost, S.D.W.; Kosakovsky Pond, S.L. Spidermonkey: Rapid detection of co-evolving sites using Bayesian graphical models. Bioinformatics 2008, 24, 1949–1950. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Frost, S.D.W.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Yohai, V.J.; Stahel, W.A.; Zamar, R.H. A procedure for robust estimation and inference in linear regression. In Directions in Robust Statistics and Diagnostics; Springer: Berlin, Germany, 1991; pp. 365–374. [Google Scholar]
- Gao, Y.; Zhao, H.; Jin, Y.; Xu, X.; Han, G.Z. Extent and evolution of gene duplication in DNA viruses. Virus Res. 2017, 240, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Frederico, L.A.; Kunkel, T.A.; Shaw, B.R. A sensitive genetic assay for the detection of cytosine deamination: Determination of rate constants and the activation energy. Biochemistry 1990, 29, 2532–2537. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. The Origins of Genome Architecture; Sinauer Associates: Sunderland, MA, USA, 2007. [Google Scholar]
- Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R. From molecular genetics to phylodynamics: Evolutionary relevance of mutation rates across viruses. PLoS Pathog. 2012, 8, e1002685. [Google Scholar] [CrossRef]
- Robinson, C.M.; Seto, D.; Jones, M.S.; Dyer, D.W.; Chodosh, J. Molecular evolution of human species D adenoviruses. Infect. Genet. Evol. 2011, 11, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Robertson, D.L.; Lovell, S.C. Constraints from protein structure and intra-molecular coevolution influence the fitness of HIV-1 recombinants. Virology 2014, 454–455, 34–39. [Google Scholar] [CrossRef]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef]
- De Paepe, M.; Taddei, F. Viruses’ life history: Towards a mechanistic basis of a trade-off between survival and reproduction among phages. PLoS Biol. 2006, 4, e193. [Google Scholar] [CrossRef]
- García-Villada, L.; Drake, J.W. Experimental selection reveals a trade-off between fecundity and lifespan in the coliphage Qß. Open Biol. 2013, 3, 130043. [Google Scholar] [CrossRef] [PubMed]
- Weinreich, D.M.; Delaney, N.F.; Depristo, M.A.; Hartl, D.L. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 2006, 312, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Pedruzzi, G.; Barlukova, A.; Rouzine, I.M. Evolutionary footprint of epistasis. PLoS Comput. Biol. 2018, 14, e1006426. [Google Scholar] [CrossRef] [PubMed]
- Sandie, R.; Aris-Brosou, S. Predicting the emergence of H3N2 influenza viruses reveals contrasted modes of evolution of HA and NA antigens. J. Mol. Evol. 2014, 78, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nun, M.; Riley, P.; Turtle, J.; Bacon, D.P.; Riley, S. Forecasting national and regional influenza-like illness for the USA. PLoS Comput. Biol. 2019, 15, e1007013. [Google Scholar] [CrossRef] [PubMed]
- Neverov, A.D.; Kryazhimskiy, S.; Plotkin, J.B.; Bazykin, G.A. Coordinated evolution of Influenza A surface proteins. PLoS Genet. 2015, 11, e1005404. [Google Scholar] [CrossRef] [PubMed]
- Ashenberg, O.; Padmakumar, J.; Doud, M.B.; Bloom, J.D. Deep mutational scanning identifies sites in influenza nucleoprotein that affect viral inhibition by MxA. PLoS Pathog. 2017, 13, e1006288. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aris-Brosou, S.; Parent, L.; Ibeh, N. Viral Long-Term Evolutionary Strategies Favor Stability over Proliferation. Viruses 2019, 11, 677. https://doi.org/10.3390/v11080677
Aris-Brosou S, Parent L, Ibeh N. Viral Long-Term Evolutionary Strategies Favor Stability over Proliferation. Viruses. 2019; 11(8):677. https://doi.org/10.3390/v11080677
Chicago/Turabian StyleAris-Brosou, Stéphane, Louis Parent, and Neke Ibeh. 2019. "Viral Long-Term Evolutionary Strategies Favor Stability over Proliferation" Viruses 11, no. 8: 677. https://doi.org/10.3390/v11080677
APA StyleAris-Brosou, S., Parent, L., & Ibeh, N. (2019). Viral Long-Term Evolutionary Strategies Favor Stability over Proliferation. Viruses, 11(8), 677. https://doi.org/10.3390/v11080677