Measles Vaccine Virus RNA in Children More Than 100 Days after Vaccination



Abstract
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Centers for Disease Control and Prevention. Measles Transmission. Available online: https://www.cdc.gov/measles/about/transmission.html (accessed on 23 June 2019).
- World Health Organization. Measles vaccines: WHO position paper. Wkly. Epidemiol. Rec. 2017, 92, 205–228. [Google Scholar]
- Perry, R.T.; Halsey, N.A. The clinical significance of measles: A review. J. Infect. Dis. 2004, 189 (Suppl. 1), S4–S16. [Google Scholar] [PubMed]
- Mina, M.J.; Metcalf, C.J.; de Swart, R.L.; Osterhaus, A.D.; Grenfell, B.T. Long-term measles-induced immunomodulation increases overall childhood infectious disease mortality. Science 2015, 348, 694–699. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Measles: Factsheet. Available online: http://www.who.int/news-room/fact-sheets/detail/measles (accessed on 23 June 2019).
- Rota, P.A.; Brown, K.; Mankertz, A.; Santibanez, S.; Shulga, S.; Muller, C.P.; Hubschen, J.M.; Siqueira, M.; Beirnes, J.; Ahmed, H.; et al. Global distribution of measles genotypes and measles molecular epidemiology. J. Infect. Dis. 2011, 204 (Suppl. 1), S514–S523. [Google Scholar] [CrossRef] [PubMed]
- Heywood, A.E.; Gidding, H.F.; Riddell, M.A.; McIntyre, P.B.; MacIntyre, C.R.; Kelly, H.A. Elimination of endemic measles transmission in Australia. Bull. World Health Organ. 2009, 87, 64–71. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Third Annual Meeting of the Regional Verification Commission for Measles Elimination in the Western Pacific: Meeting Report. Available online: https://apps.who.int/iris/bitstream/handle/10665/208616/RS_2014_GE_04_KOR_eng.pdf (accessed on 23 June 2019).
- Dabbagh, A.; Laws, R.L.; Steulet, C.; Dumolard, L.; Mulders, M.N.; Kretsinger, K.; Alexander, J.P.; Rota, P.A.; Goodson, J.L. Progress toward regional measles elimination—Worldwide, 2000–2017. Morb. Mortal. Wkly. Rep. 2018, 67, 1323–1329. [Google Scholar] [CrossRef]
- Gastanaduy, P.A.; Banerjee, E.; DeBolt, C.; Bravo-Alcantara, P.; Samad, S.A.; Pastor, D.; Rota, P.A.; Patel, M.; Crowcroft, N.S.; Durrheim, D.N. Public health responses during measles outbreaks in elimination settings: Strategies and challenges. Hum. Vaccines Immunother. 2018, 14, 2222–2238. [Google Scholar] [CrossRef]
- National Health and Medical Research Council. The Australian Research Council and Universities Australia. Commonwealth of Australia. National Statement on Ethical Conduct in Human Research 2007 (Updated 2018). Available online: https://www.nhmrc.gov.au/about-us/publications/national-statement-ethical-conduct-human-research-2007-updated-2018 (accessed on 23 June 2019).
- McMahon, J.; Northill, J.; Finger, M.; Lyon, M.; Lambert, S.; Mackay, I. Laboratory methods supporting measles surveillance in Queensland, Australia, 2010–2017. BioRxiv 2018. [Google Scholar] [CrossRef]
- Chibo, D.; Birch, C.J.; Rota, P.A.; Catton, M.G. Molecular characterization of measles viruses isolated in Victoria, Australia, between 1973 and 1998. J. Gen. Virol. 2000, 81, 2511–2518. [Google Scholar] [CrossRef]
- Tran, T.; Kostecki, R.; Catton, M.; Druce, J. Utility of a stressed single nucleotide polymorphism (SNP) real-time PCR assay for rapid identification of measles vaccine strains in patient samples. J. Clin. Microbiol. 2018, 56, e00360-18. [Google Scholar] [CrossRef]
- Allen, I.V.; McQuaid, S.; Penalva, R.; Ludlow, M.; Duprex, W.P.; Rima, B.K. Macrophages and dendritic cells are the predominant cells infected in measles in humans. mSphere 2018, 3, e00570-17. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.E.; Lin, W.W.; Nelson, A.N. Understanding the causes and consequences of measles virus persistence. F1000Res 2018, 7, 237. [Google Scholar] [CrossRef] [PubMed]
- Naaman, H.; Rabinski, T.; Yizhak, A.; Mizrahi, S.; Avni, Y.S.; Taube, R.; Rager, B.; Weinstein, Y.; Rall, G.; Gopas, J.; et al. Measles virus persistent infection of human induced pluripotent stem cells. Cell. Reprogram. 2018, 20, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.E.; Griffin, D.E. Within host RNA virus persistence: Mechanisms and consequences. Curr. Opin. Virol. 2017, 23, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Riddell, M.A.; Moss, W.J.; Hauer, D.; Monze, M.; Griffin, D.E. Slow clearance of measles virus RNA after acute infection. J. Clin. Virol. 2007, 39, 312–317. [Google Scholar] [CrossRef]
- Lin, W.H.; Kouyos, R.D.; Adams, R.J.; Grenfell, B.T.; Griffin, D.E. Prolonged persistence of measles virus RNA is characteristic of primary infection dynamics. Proc. Natl. Acad. Sci. USA 2012, 109, 14989–14994. [Google Scholar] [CrossRef]
- Fisher, D.L.; Defres, S.; Solomon, T. Measles-induced encephalitis. QJM 2015, 108, 177–182. [Google Scholar] [CrossRef]
- Cattaneo, R.; Schmid, A.; Spielhofer, P.; Kaelin, K.; Baczko, K.; Ter Meulen, V.; Pardowitz, J.; Flanagan, S.; Rima, B.K.; Udem, S.A.; et al. Mutated and hypermutated genes of persistent measles viruses which caused lethal human brain diseases. Virology 1989, 173, 415–425. [Google Scholar] [CrossRef]
- Hornig, M.; Briese, T.; Buie, T.; Bauman, M.L.; Lauwers, G.; Siemetzki, U.; Hummel, K.; Rota, P.A.; Bellini, W.J.; O’Leary, J.J.; et al. Lack of association between measles virus vaccine and autism with enteropathy: A case-control study. PLoS ONE 2008, 3, e3140. [Google Scholar] [CrossRef]
- Sonoda, S.; Nakayama, T. Detection of measles virus genome in lymphocytes from asymptomatic healthy children. J. Med. Virol. 2001, 65, 381–387. [Google Scholar] [CrossRef]
- Sonoda, S.; Kitahara, M.; Nakayama, T. Detection of measles virus genome in bone-marrow aspirates from adults. J. Gen. Virol. 2002, 83, 2485–2488. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Kohso, K.; Nishimura, A.; Tatsuno, Y.; Homma, M.; Hotta, H. Detection of measles virus mRNA from autopsied human tissues. J. Clin. Microbiol. 1998, 36, 299–301. [Google Scholar] [PubMed]
- Su, J.R.; Ng, C.; Lewis, P.W.; Cano, M.V. Adverse events after vaccination among HIV-positive persons, 1990–2016. PLoS ONE 2018, 13, e0199229. [Google Scholar] [CrossRef] [PubMed]
- Bitnun, A.; Shannon, P.; Durward, A.; Rota, P.A.; Bellini, W.J.; Graham, C.; Wang, E.; Ford-Jones, E.L.; Cox, P.; Becker, L.; et al. Measles inclusion-body encephalitis caused by the vaccine strain of measles virus. Clin. Infect. Dis. 1999, 29, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.J.; Mohamad, S.M.; Young, D.F.; Skelton, A.J.; Leahy, T.R.; Munday, D.C.; Butler, K.M.; Morfopoulou, S.; Brown, J.R.; Hubank, M.; et al. Human IFNAR2 deficiency: Lessons for antiviral immunity. Sci. Transl. Med. 2015, 7, 307ra154. [Google Scholar] [CrossRef]
- Berggren, K.L.; Tharp, M.; Boyer, K.M. Vaccine-associated “wild-type” measles. Pediatr. Dermatol. 2005, 22, 130–132. [Google Scholar] [CrossRef]
- Rota, P.A.; Khan, A.S.; Durigon, E.; Yuran, T.; Villamarzo, Y.S.; Bellini, W.J. Detection of measles virus RNA in urine specimens from vaccine recipients. J. Clin. Microbiol. 1995, 33, 2485–2488. [Google Scholar]
- Tramuto, F.; Dones, P.; D’Angelo, C.; Casuccio, N.; Vitale, F. Post-vaccine measles in a child with concomitant influenza, Sicily, Italy, March 2015. Eurosurveillance 2015, 20, 21134. [Google Scholar] [CrossRef][Green Version]
- Murti, M.; Krajden, M.; Petric, M.; Hiebert, J.; Hemming, F.; Hefford, B.; Bigham, M.; Van Buynder, P. Case of vaccine-associated measles five weeks post-immunisation, British Columbia, Canada, October 2013. Eurosurveillance 2013, 18, 20649. [Google Scholar] [CrossRef]
- Jenkin, G.A.; Chibo, D.; Kelly, H.A.; Catton, M.G.; Lynch, P.A. What is the cause of a rash after measles-mumps-rubella vaccination? Med. J. Aust. 1999, 171, 194–195. [Google Scholar] [CrossRef]
- Greenwood, K.P.; Hafiz, R.; Ware, R.S.; Lambert, S.B. A systematic review of human-to-human transmission of measles vaccine virus. Vaccine 2016, 34, 2531–2536. [Google Scholar] [CrossRef] [PubMed]



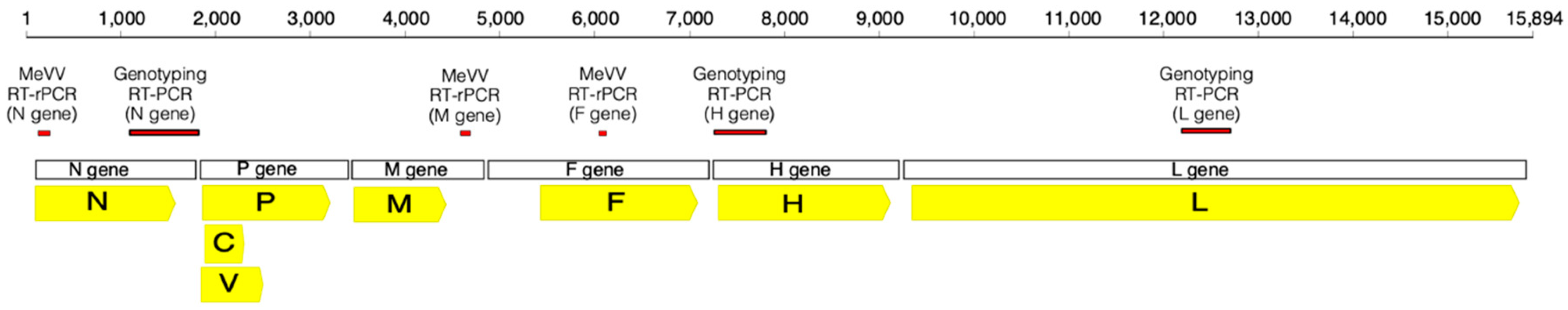{kind=link}
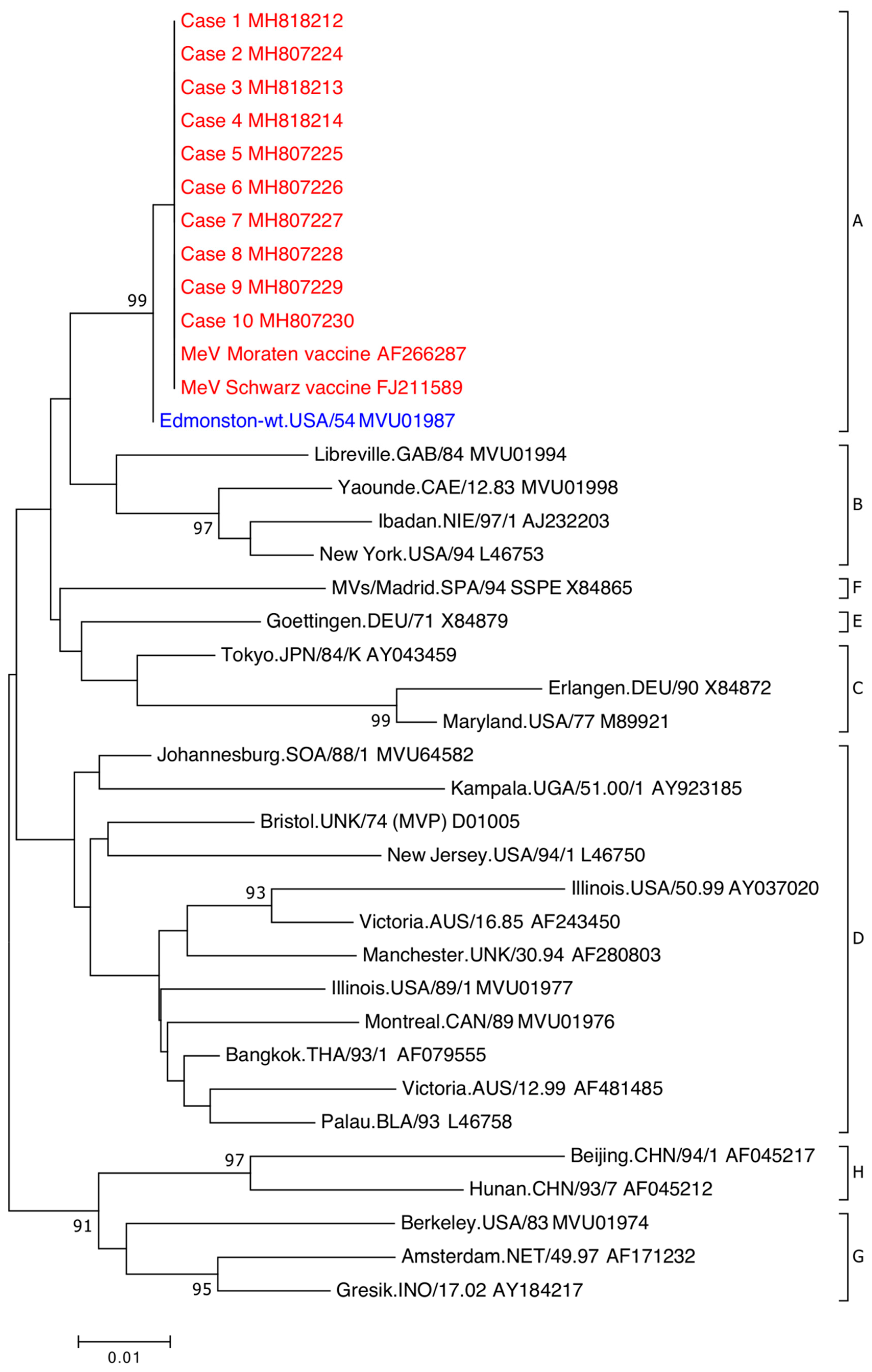{kind=link}
| Case | Age, Sex | Days Post-Vaccination | Swab Site | Request Notes | Vaccine § | Concurrent Detection/s ǁ | Designated Genotype (N Gene RT-PCR ‡) |
|---|---|---|---|---|---|---|---|
| 1 | 23mo, F | 218 | NP | Third day high fevers, rash appearing | MCV1 Priorix | HMPV | Genotype A |
| 2 | 17mo, F | 142 | NP | Query measles | MCV1 M-M-R II | NT | Genotype A |
| 3 | 25mo, M | 345 | Nasal | In Europe two weeks before illness | MCV1 Priorix | RSV | Genotype A |
| 4 | 30mo, F | 548 | NP | Viral infection, lower lobe consolidation | MCV1 Priorix | AdV | Genotype A |
| 5 | 16mo, F | 125 | Swab * | Rash, fever | MCV1 M-M-R II | RSV HPIV-3 | Genotype A |
| 6 | 16mo, M | 147 | NP | No notes | MCV1 M-M-R II | NT | Genotype A |
| 7 | 33mo, M | 471 | NP | No notes | MCV2 Priorix-tetra | NT | Genotype A |
| 8 | 15mo, M | 101 | NP | Non-itchy, red throat, whole-body rash, possible Koplik spots | MCV1 Priorix | ND | Genotype A |
| 9 | 16mo, F | 110 | Nasal | Fever, rash. Rubella contact | MCV1 M-M-R II | NT | Genotype A |
| 10 | 17mo, F | 139 | Swab * | Viral rash on face | MCV1 M-M-R II | AdV | Genotype A |
| 11 | 45mo, M | 784 | NP | No notes | MCV2 Priorix-tetra | NT | Insufficient RNA |
| Case | Days Post-Vaccination | MeV F Gene RT-rPCR (CT Value) † | MeVV RT-rPCR (CT Value) † | MeV N Gene RT-rPCR (CT Value) † | N Gene RT-PCR ‡ | L Gene RT-PCR ‡ | H Gene RT-PCR ‡ | Designated Genotype (N Gene RT-PCR ‡∂) | Urine MeV F Gene RT-rPCR |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 218 | 33.21 32.86 | 34.12 33.29 | NT | DET | DET | DET | Genotype A | ND |
| 2 | 142 | 38.20 37.62 | 39.30 ND | 39.14 38.10 | DET | DET | ND | Genotype A | NS |
| 3 | 345 | 37.11 36.87 | 38.11 ND | 38.60 37.33 | DET | DETα | DET | Genotype A | ND |
| 4 | 548 | 36.40 37.24 | ND * 38.93 * | 38.93 ND | DET | DET | DET | Genotype A | NS |
| 5 | 125 | 34.09 34.22 | 36.64 36.50 | 36.98 36.62 | DET | DET | DET | Genotype A | NS |
| 6 | 147 | 33.90 ND | 35.22 ND | 36.78 36.65 | DET | DET | DET | Genotype A | ND |
| 7 | 471 | 37.47 38.51 | 39.13 39.46 | ND 39.21 | DET | ND | DETα | Genotype A | ND |
| 8 | 101 | 39.62 ND | 38.82 ND | 33.58 33.46 | DET | DET α | DET | Genotype A | NS |
| 9 | 110 | 32.72 33.37 | 35.34 35.28 | 35.82 34.71 | DET | DET | DET | Genotype A | NS |
| 10 | 139 | 38.33 ND | 39.30 ND | NT | DET | DET | DET | Genotype A | NS |
| 11 | 784 | 39.62 ND | ND 38.82 | 39.41 39.88 | DETα | DET | ND | Insufficient RNA | NS |
| DAYS SINCE LAST MCV * | NUMBER OF MEVV CASES |
|---|---|
| 0–19 | 106 |
| 20–39 | 10 |
| 40–59 | 5 |
| 60–79 | 4 |
| 80–100 | 3 |
| >100 | 11 |
| UNKNOWN | 2 |
| Case | PCR Testing Performed | Detections |
|---|---|---|
| 1 | RSV, IFAV, IFBV, HPIV-1, HPIV-2, HPIV-3, HMPV, RV, AdV | HMPV |
| 2 | Not tested for other viruses | NT |
| 3 | RSV, IFAV, IFBV, HPIV-1, HPIV-2, HPIV-3, HMPV, RV, AdV | RSV |
| 4 | RSV, IFAV, IFBV, HPIV-1, HPIV-2, HPIV-3, HMPV, RV, AdV | AdV |
| 5 | RSV, IFAV, IFBV, HPIV-1, HPIV-2, HPIV-3, HMPV, RV, AdV | RSV, HPIV-3 |
| 6 | RSV, IFAV, IFBV, HPIV-1, HPIV-2, HPIV-3, HMPV, RV, AdV | ND |
| 7 | Not tested for other viruses | NT |
| 8 | RUBV *, RSV, IFAV, IFBV, HPIV-1, HPIV-2, HPIV-3, HMPV, RV, AdV | ND |
| 9 | RUBV * | ND |
| 10 | RSV, IFAV, IFBV, HPIV-1, HPIV-2, HPIV-3, HMPV, RV, AdV | AdV |
| 11 | Not tested for other viruses | NT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McMahon, J.; Mackay, I.M.; Lambert, S.B. Measles Vaccine Virus RNA in Children More Than 100 Days after Vaccination. Viruses 2019, 11, 636. https://doi.org/10.3390/v11070636
McMahon J, Mackay IM, Lambert SB. Measles Vaccine Virus RNA in Children More Than 100 Days after Vaccination. Viruses. 2019; 11(7):636. https://doi.org/10.3390/v11070636
Chicago/Turabian StyleMcMahon, Jamie, Ian M Mackay, and Stephen B Lambert. 2019. "Measles Vaccine Virus RNA in Children More Than 100 Days after Vaccination" Viruses 11, no. 7: 636. https://doi.org/10.3390/v11070636
APA StyleMcMahon, J., Mackay, I. M., & Lambert, S. B. (2019). Measles Vaccine Virus RNA in Children More Than 100 Days after Vaccination. Viruses, 11(7), 636. https://doi.org/10.3390/v11070636