Two Phages of the Genera Felixunavirus Subjected to 12 Hour Challenge on Salmonella Infantis Showed Distinct Genotypic and Phenotypic Changes


 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Characterization of Host Bacteria and Bacteriophages
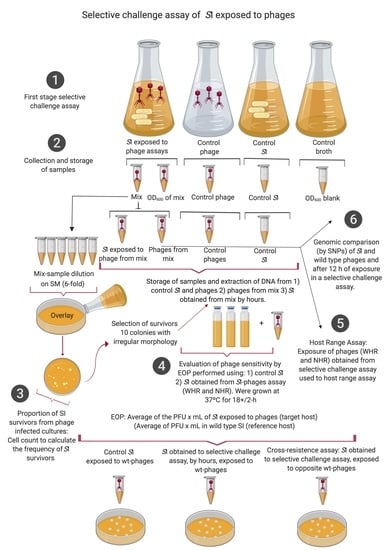
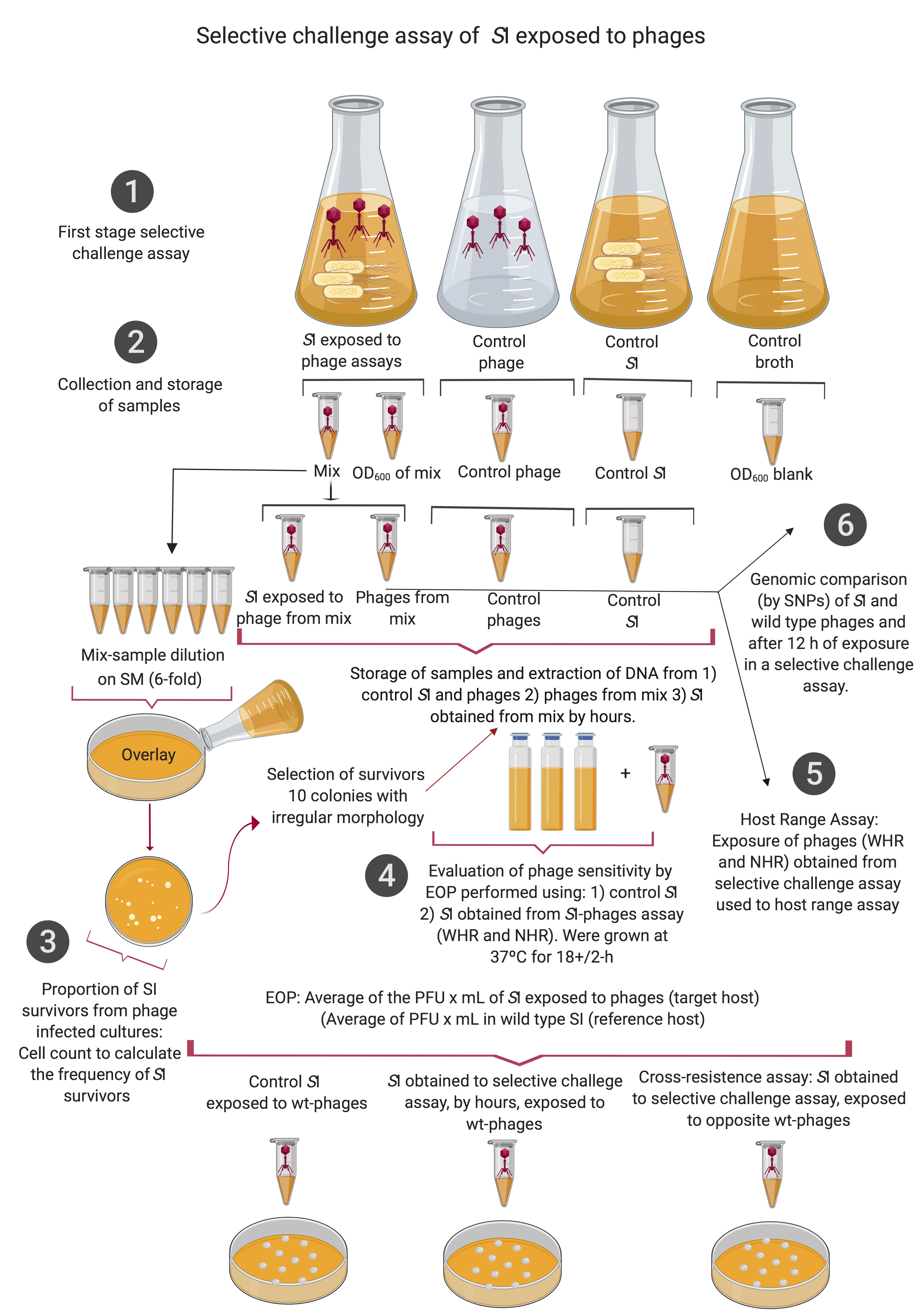
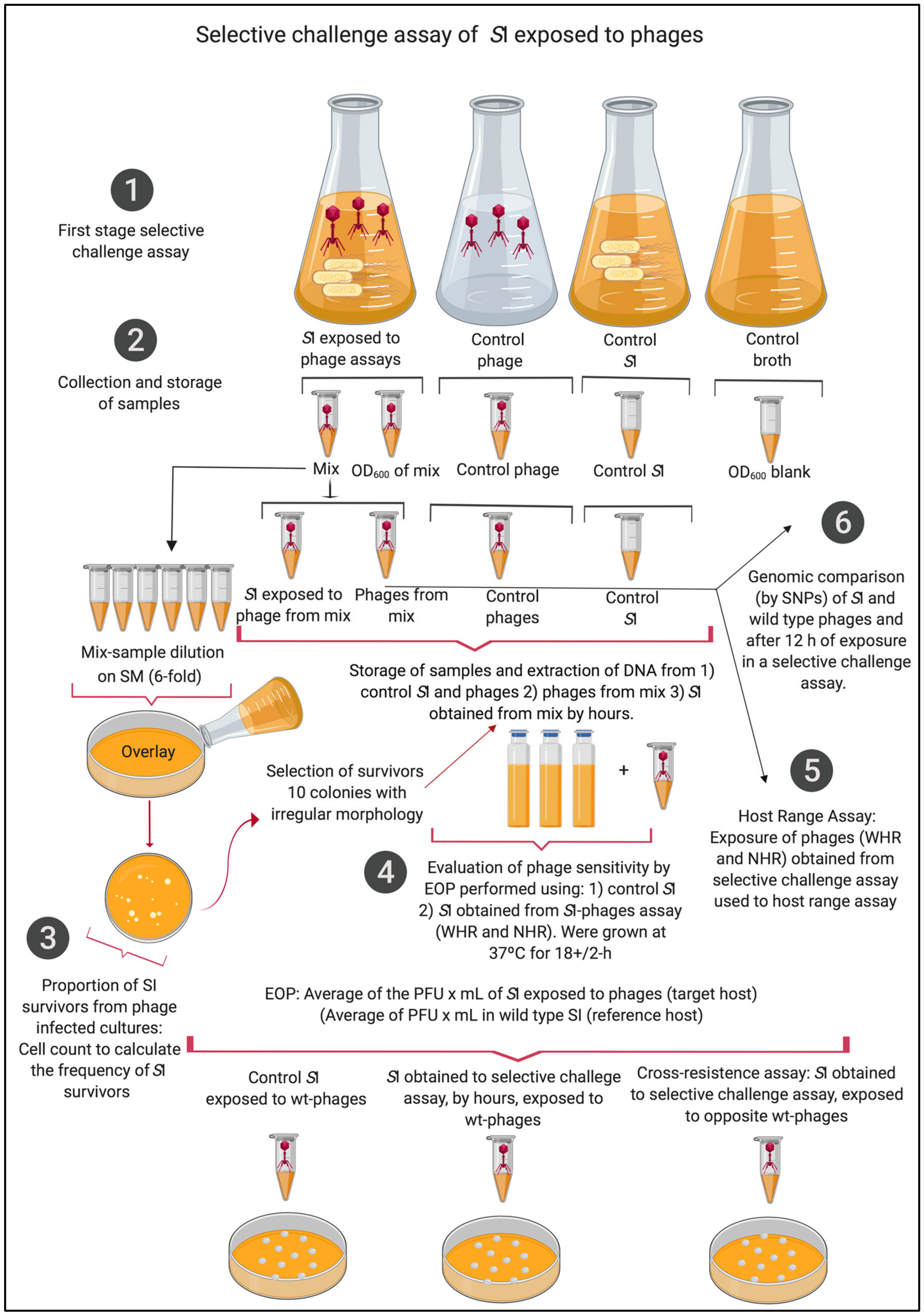
2.2. Selective Challenge Assay of SI Exposed to WHR and NHR Phages
2.3. Statistical Analysis
2.4. Evaluation of Phage Sensitivity by Efficiency of Plating on Isolated SI Survivors
2.5. Efficiency of Plating on SI Survivors Obtained from Cross-Resistance Assays
2.6. Genotypic Evaluation of Changes in SI and Phages Obtained from the Selective Challenge Assay
2.7. Phenotypic Evaluation of Changes in the Host Range of WHR and NHR Phages, Obtained from the Selective Challenge Assay
3. Results and Discussion
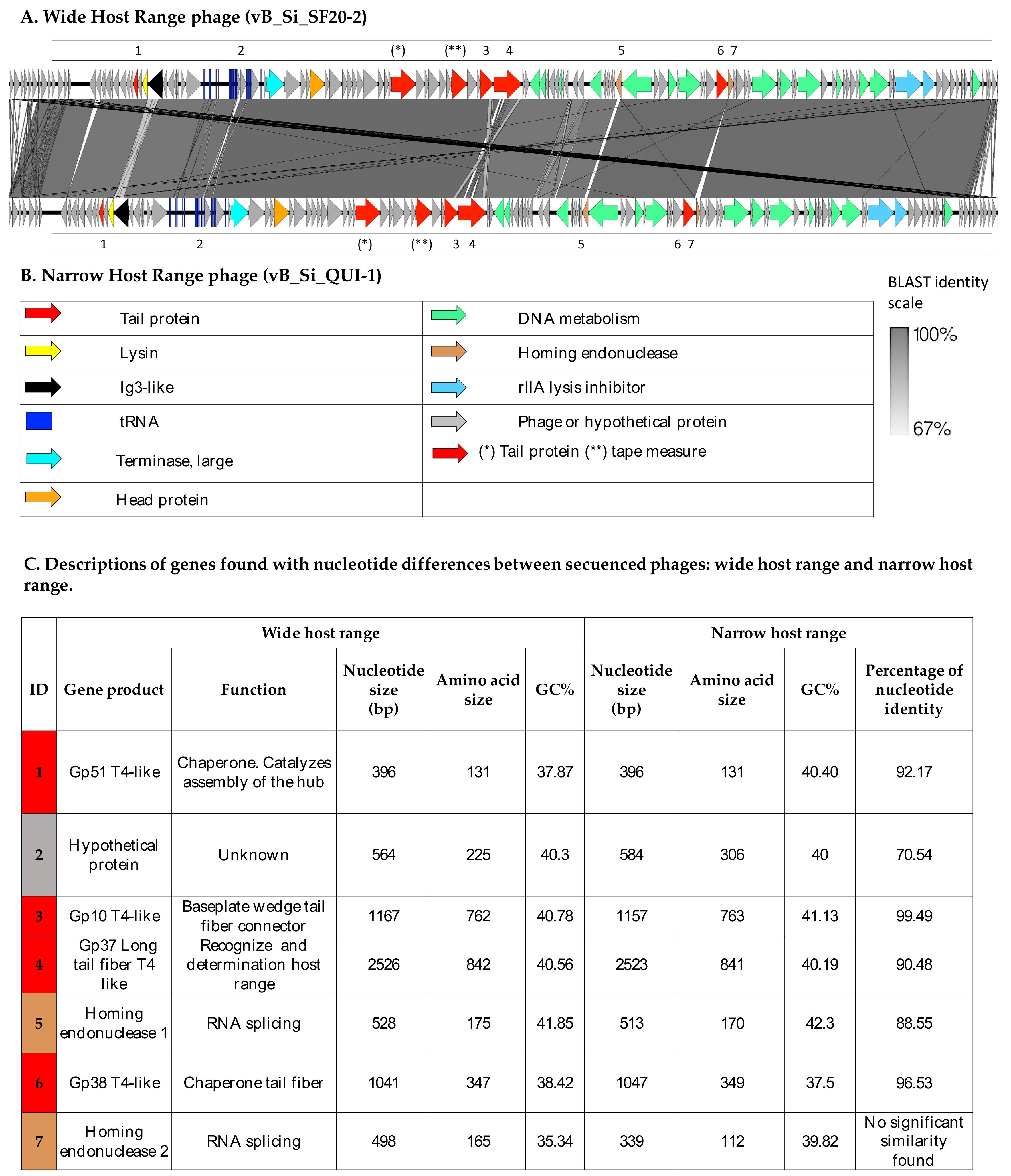
3.1. Two Highly Similar Phages Present Distinct Genotypic and Phenotypic Characteristics
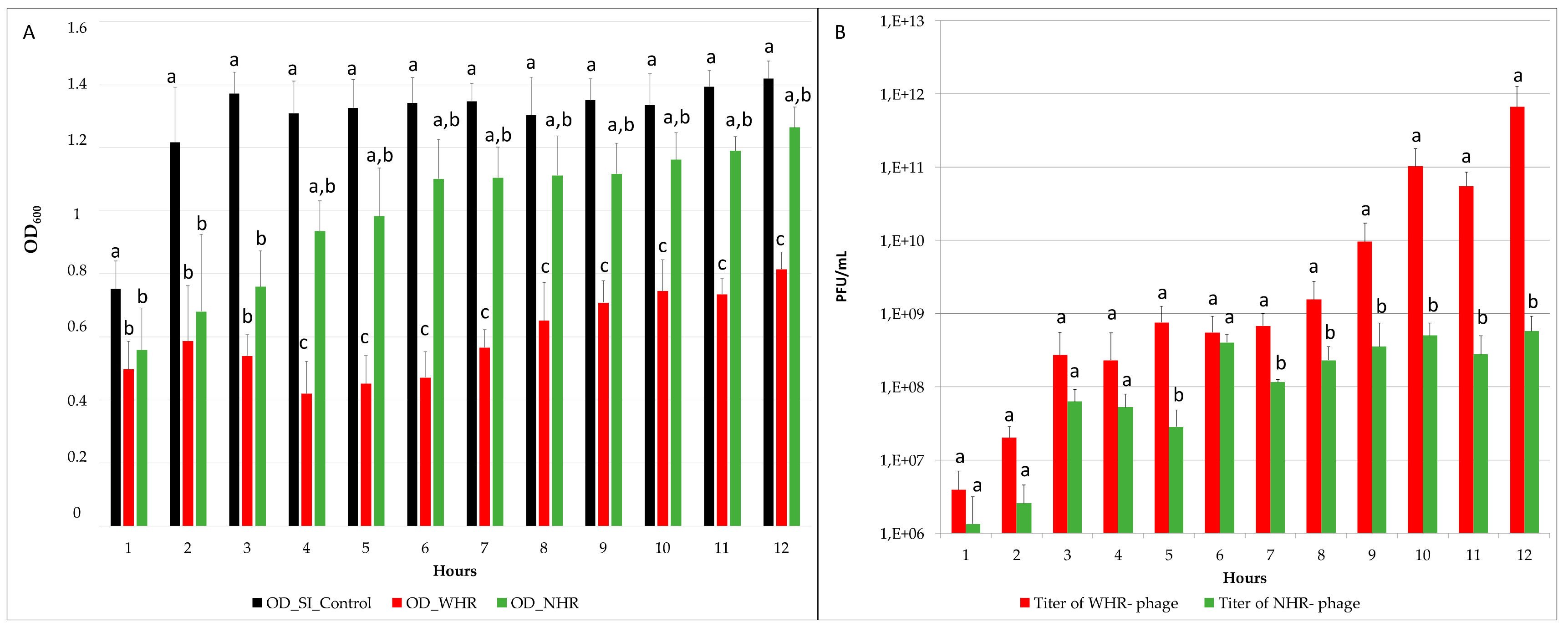
3.2. The Two Phages Showed Significantly Different Effects on SI OD and Titer Post-Infection
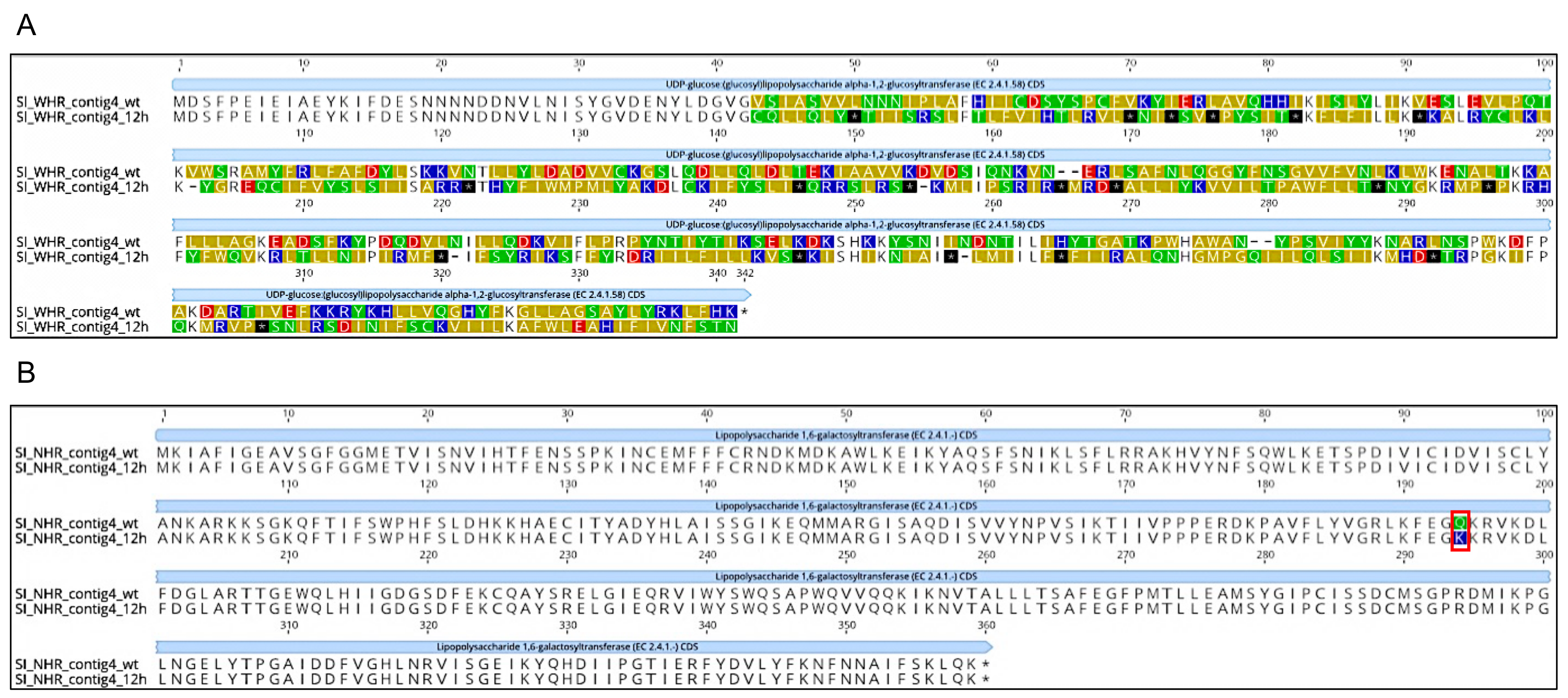
3.3. SI and Phages Generated Different Genotypic Responses to the Selective Challenge Assays
3.4. SI and Phages Showed Different Phenotypic Responses to Selective Challenge Assays
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- European Food Safety Authority (EFSA) and European Centre for Disease Prevention and Control. The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2017. EFSA J. 2018, 16, 22–64. [Google Scholar]
- Center for Disease Control (CDC) and Prevention. National Enteric Disease Surveillance: Salmonella Annual Summary. Available online: https://www.cdc.gov/nationalsurveillance/salmonella-surveillance.html (accessed on 20 April 2019).
- Center for Disease Control (CDC) and Prevention Outbreak of Multidrug-Resistant Salmonella Infections Linked to Raw Chicken Products. Available online: https://www.cdc.gov/salmonella/infantis-10-18/index.html (accessed on 20 April 2019).
- Fernández, J.; Guerra, B.; Rodicio, M. Resistance to Carbapenems in Non-Typhoidal Salmonella enterica Serovars from Humans, Animals and Food. Vet. Sci. 2018, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Carfora, V.; Alba, P.; Leekitcharoenphon, P.; Ballarò, D.; Cordaro, G.; Di Matteo, P.; Donati, V.; Ianzano, A.; Iurescia, M.; Stravino, F.; et al. Colistin resistance mediated by mcr-1 in ESBL-producing, multidrug resistant Salmonella Infantis in broiler chicken industry, Italy (2016–2017). Front. Microbiol. 2018, 9, 1880–1886. [Google Scholar] [CrossRef] [PubMed]
- Borowiak, M.; Fischer, J.; Baumann, B.; Hammerl, J.A.; Szabo, I.; Malorny, B. Complete Genome Sequence of a VIM-1-Producing Salmonella enterica subsp. enterica Serovar Infantis Isolate Derived from Minced Pork Meat. Genome Announc. 2018, 6, 318–327. [Google Scholar]
- Franco, A.; Leekitcharoenphon, P.; Feltrin, F.; Alba, P.; Cordaro, G.; Iurescia, M.; Tolli, R.; D’Incau, M.; Staffolani, M.; Di Giannatale, E.; et al. Emergence of a Clonal Lineage of Multidrug-Resistant ESBL-Producing Salmonella Infantis Transmitted from Broilers and Broiler Meat to Humans in Italy between 2011 and 2014. PLoS ONE 2015, 10, e0144802. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Peighambari, S.M.; Svendsen, C.A.; Cavaco, L.M.; Agersø, Y.; Hendriksen, R.S. Molecular clonality and antimicrobial resistance in Salmonella enterica serovars Enteritidis and Infantis from broilers in three Northern regions of Iran. BMC Vet. Res. 2013, 48, 1746–1761. [Google Scholar]
- Hudson, J.A.; Billington, C.; Carey-Smith, G.; Greening, G. Bacteriophages as Biocontrol. J. Food Prot. 2005, 68, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Hagens, S.; Loessner, M. Bacteriophage for Biocontrol of Foodborne Pathogens: Calculations and Considerations. Curr. Pharm. Biotechnol. 2010, 11, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Berchieri, A., Jr.; Lovell, M.A.; Barrow, P.A. The activity in the chicken alimentary tract of bacteriophages lytic for Salmonella typhimurium. Res. Microbiol. 1991, 142, 541–549. [Google Scholar] [CrossRef]
- Anany, H.; Chen, W.; Pelton, R.; Griffiths, M.W. Biocontrol of Listeria monocytogenes and Escherichia coli O157:H7 in Meat by Using Phages Immobilized on Modified Cellulose Membranes. Appl. Environ. Microbiol. 2011, 77, 6379–6387. [Google Scholar] [CrossRef]
- Greer, G.G. Effects of Phage Concentration, Bacterial Density, and Temperature on Phage Control of Beef Spoilage. J. Food Sci. 1988, 53, 1226–1227. [Google Scholar] [CrossRef]
- Luria, S.E.; Delbrück, M. Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics 1943, 28, 491–511. [Google Scholar] [PubMed]
- Hyman, P.; Abedon, S.T. Bacteriophage Host Range and Bacterial Resistance. Adv. Appl. Microbiol. 2010, 70, 217–248. [Google Scholar] [PubMed]
- Denes, T.; Den Bakker, H.C.; Tokman, J.I.; Guldimann, C.; Wiedmann, M. Selection and characterization of phage-resistant mutant strains of Listeria monocytogenes reveal host genes linked to phage adsorption. Appl. Environ. Microbiol. 2015, 81, 4295–4305. [Google Scholar] [CrossRef] [PubMed]
- Holmfeldt, K.; Solonenko, N.; Howard-Varona, C.; Moreno, M.; Malmstrom, R.R.; Blow, M.J.; Sullivan, M.B. Large-scale maps of variable infection efficiencies in aquatic Bacteroidetes phage-host model systems. Environ. Microbiol. 2016, 18, 3949–3961. [Google Scholar] [CrossRef]
- Yuan, Y.; Peng, Q.; Zhang, S.; Liu, T.; Yang, S.; Yu, Q.; Wu, Y.; Gao, M. Phage reduce stability for regaining infectivity during antagonistic coevolution with host bacterium. Viruses 2019, 2, 118. [Google Scholar] [CrossRef] [PubMed]
- Friman, V.P.; Hiltunen, T.; Jalasvuori, M.; Lindstedt, C.; Laanto, E.; Örmälä, A.M.; Laakso, J.; Mappes, J.; Bamford, J.K.H. High temperature and bacteriophages can indirectly select for bacterial pathogenicity in environmental reservoirs. PLoS ONE 2011, 6, e17651. [Google Scholar] [CrossRef]
- Laanto, E.; Bamford, J.K.H.; Laakso, J.; Sundberg, L.R. Phage-Driven Loss of Virulence in a Fish Pathogenic Bacterium. PLoS ONE 2012, 12, e53157. [Google Scholar] [CrossRef]
- Heller, K.J. Molecular interaction between bacteriophage and the gram-negative cell envelope. Arch. Microbiol. 1992, 158, 235–248. [Google Scholar] [CrossRef]
- Tétart, F.; Repoila, F.; Monod, C.; Krisch, H.M. Bacteriophage T4 host range is expanded by duplications of a small domain of the tail fiber adhesin. J. Mol. Biol. 1996, 258, 726–731. [Google Scholar] [CrossRef]
- Liu, M.; Gingery, M.; Doulatov, S.R.; Liu, Y.; Hodes, A.; Baker, S.; Davis, P.; Simmonds, M.; Churcher, C.; Mungall, K.; et al. Genomic and Genetic Analysis of Bordetella Bacteriophages Encoding Reverse Transcriptase-Mediated Tropism-Switching Cassettes. J. Bacteriol. 2004, 186, 1503–1517. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, D.; Buettner, F.F.; Browning, C.; Nazarov, S.; Rabsch, W.; Bethe, A.; Oberbeck, A.; Bowman, V.D.; Stummeyer, K.; Mühlenhoff, M.; et al. A multivalent adsorption apparatus explains the broad host range of phage phi92: A comprehensive genomic and structural analysis. J. Virol. 2012, 86, 10384–10398. [Google Scholar] [CrossRef] [PubMed]
- Connerton, P.L.; Loc Carrillo, C.M.; Swift, C.; Dillon, E.; Scott, A.; Rees, C.E.; Dodd, C.E.; Frost, J.; Connerton, I.F. Longitudinal study of Campylobacter jejuni bacteriophages and their hosts from broiler chickens. Appl. Environ. Microbiol. 2004, 70, 3877–3883. [Google Scholar] [CrossRef] [PubMed]
- Fister, S.; Robben, C.; Witte, A.K.; Schoder, D.; Wagner, M.; Rossmanith, P. Influence of Environmental Factors on Phage—Bacteria Interaction and on the Efficacy and Infectivity of Phage P100. Front. Microbiol. 2016, 7, 1152–1165. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Wittmann, J.; Kuhn, J.H.; Turner, D.; Sullivan, M.B.; Dutilh, B.E.; Jang, H.B.; van Zyl, L.J.; Klumpp, J.; Lobocka, M.; et al. Taxonomy of prokaryotic viruses: 2017 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2018, 163, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Leiman, P.G.; Shneider, M.M. Contractile Tail Machines of Bacteriophages. In Viral Molecular Machines; Rossmann, M., Rao, V., Eds.; Springer: Boston, MA, USA, 2012; pp. 93–114. ISBN 978-1-4614-0980-9. Available online: https://link.springer.com/chapter/10.1007%2F978-1-4614-0980-9_5#citeas (accessed on 2 March 2019).
- Welkos, S.; Schreiber, M.; Baer, H. Identification of Salmonella with the 0–1 Bacteriophage. Appl. Microbiol. 1974, 28, 618–622. [Google Scholar] [PubMed]
- Marti, R.; Zurfluh, K.; Hagens, S.; Pianezzi, J.; Klumpp, J.; Loessner, M.J. Long tail fibres of the novel broad-host-range T-even bacteriophage S16 specifically recognize Salmonella OmpC. Mol. Microbiol. 2013, 87, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Koskella, B.; Meaden, S. Understanding bacteriophage specificity in natural microbial communities. Viruses 2013, 5, 806–823. [Google Scholar] [CrossRef]
- Ross, A.; Ward, S.; Hyman, P. More is better: Selecting for broad host range bacteriophages. Front. Microbiol. 2016, 7, 1352. [Google Scholar] [CrossRef]
- Poullain, V.; Gandon, S.; Brockhurst, M.A.; Buckling, A.; Hochberg, M.E. The evolution of specificity in evolving and coevolving antagonistic interactions between a bacteria and its phage. Evolution 2008, 62, 1–11. [Google Scholar] [CrossRef]
- Takeuchi, I.; Osada, K.; Azam, A.H.; Asakawa, H.; Miyanaga, K.; Tanji, Y. The Presence of Two Receptor-Binding Proteins Contributes to the Wide Host Range of Staphylococcal Twort-Like Phages. Appl. Environ. Microbiol. 2016, 82, 5763–5774. [Google Scholar] [CrossRef] [PubMed]
- Born, Y.; Fieseler, L.; Marazzi, J.; Lurz, R.; Duffy, B.; Loessner, M.J. Novel Virulent and Broad-Host-Range Erwinia amylovora Bacteriophages Reveal a High Degree of Mosaicism and a Relationship to Enterobacteriaceae Phages. Appl. Environ. Microbiol. 2011, 77, 5945–5954. [Google Scholar] [CrossRef] [PubMed]
- Bielke, L.; Higgins, S.; Donoghue, A.; Donoghue, D.; Hargis, B.M. Salmonella host range of bacteriophages that infect multiple genera. Poult. Sci. 2007, 86, 2536–2540. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, L.; Abdelgader, S.A.; Yu, L.; Xu, J.; Yao, H.; Lu, C.; Zhang, W. Alterations in gp37 Expand the Host Range of a T4-Like Phage. Appl. Environ. Microbiol. 2017, 83, e01576-17. [Google Scholar] [CrossRef] [PubMed]
- Denyes, J.M.; Dunne, M.; Steiner, S.; Mittelviefhaus, M.; Weiss, A.; Schmidt, H.; Klumpp, J.; Loessner, M.J. Modified bacteriophage S16 long tail fiber proteins for rapid and specific immobilization and detection of Salmonella cells. Appl. Environ. Microbiol. 2017, 83, e00277-17. [Google Scholar] [CrossRef] [PubMed]
- Alegria-Moran, R.; Rivera, D.; Toledo, V.; Moreno-Switt, A.I.; Hamilton-West, C. First detection and characterization of Salmonella spp. in poultry and swine raised in backyard production systems in central Chile. Epidemiol. Infect. 2017, 145, 3180–3190. [Google Scholar] [CrossRef] [PubMed]
- Rivera, D.; Toledo, V.; Pillo, F.D.; Duenas, F.; Tardone, R.; Hamilton-West, C.; Vongkamjan, K.; Wiedmann, M.; Switt, A.I.M. Backyard Farms Represent a Source of Wide Host Range Salmonella Phages That Lysed the Most Common Salmonella Serovars. J. Food Prot. 2018, 81, 272–278. [Google Scholar] [CrossRef]
- Ackermann, H.-W. Basic phage electron microscopy. In Bacteriophages; Clokie, M.R., Kropinski, A.M., Eds.; Methods in Molecular Biology™; Humana Press: New York, NY, USA, 2009; Volume 501, pp. 113–126. Available online: https://link.springer.com/protocol/10.1007%2F978-1-60327-164-6_12 (accessed on 14 April 2019).
- Park, M.; Lee, J.-H.; Shin, H.; Kim, M.; Choi, J.; Kang, D.-H.; Sunggi, H.; Ryu, S. Characterization and comparative genomic analysis of a novel bacteriophage, SFP10, simultaneously inhibiting both Salmonella enterica and Escherichia coli O157:H7. Appl. Environ. Microbiol. 2012, 78, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Moreno Switt, A.I.; Orsi, R.H.; den Bakker, H.C.; Vongkamjan, K.; Altier, C.; Wiedmann, M. Genomic characterization provides new insight into Salmonella phage diversity. BMC Genom. 2013, 17, 481. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. EasyFig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Filippov, A.A.; Sergueev, K.V.; He, Y.; Huang, X.Z.; Gnade, B.T.; Mueller, A.J.; Fernandez-Prada, C.M.; Nikolich, M.P. Bacteriophage-resistant mutants in Yersinia pestis: Identification of phage receptors and attenuation for mice. PLoS ONE 2011, 6, e25486. [Google Scholar] [CrossRef]
- Costa, P.; Pereira, C.; Gomes, A.T.; Almeida, A. Efficiency of Single Phage Suspensions and Phage Cocktail in the Inactivation of Escherichia coli and Salmonella Typhimurium: An In Vitro Preliminary Study. Microorganisms 2019, 7, 94. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, G.G.; Park, J.S.; Jung, Y.H.; Kwak, H.S.; Kim, S.B.; Nam, Y.S.; Kwon, S.T. A novel multiplex PCR assay for rapid and simultaneous detection of five pathogenic bacteria: Escherichia coli O157:H7, Salmonella, Staphylococcus aureus, Listeria monocytogenes, and Vibrio parahaemolyticus. J. Food Prot. 2007, 70, 1656–1662. [Google Scholar] [CrossRef]
- Theodorsson-Norheim, E. Kruskal-Wallis test: BASIC computer program to perform nonparametric one-way analysis of variance and multiple comparisons on ranks of several independent samples. Comput. Methods Programs Biomed. 1986, 23, 57–62. [Google Scholar] [CrossRef]
- Quirk, T.J. One-Way Analysis of Variance (ANOVA). In Excel 2016 in Applied Statistics for High School Students; Excel for Statistics; Springer: Cham, Germany, 2018; pp. 169–185. [Google Scholar] [CrossRef]
- Petsong, K.; Benjakul, S.; Chaturongakul, S.; Switt, A.I.M.; Vongkamjan, K. Lysis Profiles of Salmonella Phages on Salmonella Isolates from Various Sources and Efficiency of a Phage Cocktail against S. Enteritidis and S. Typhimurium. Microorganisms 2019, 7, 100. [Google Scholar] [CrossRef]
- A Quality Control Tool for High Throughput Sequence Data. S. FASTQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 8 April 2019).
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Turner, I.; Garimella, K.V.; Iqbal, Z.; McVean, G. Integrating long-range connectivity information into de Bruijn graphs. Bioinformatics 2018, 34, 2556–2565. [Google Scholar] [CrossRef]
- Chikhi, R.; Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef]
- Whichard, J.M.; Weigt, L.A.; Borris, D.J.; Li, L.L.; Zhang, Q.; Kapur, V.; Pierson, F.W.; Lingohr, E.J.; She, Y.M.; Kropinski, A.M.; et al. Complete genomic sequence of bacteriophage Felix O1. Viruses 2010, 2, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Tolen, T.N.; Xie, Y.; Hernandez, A.C.; Everett, G.F.K. Complete Genome Sequence of Salmonella enterica Serovar Typhimurium Myophage Mushroom. Genome Announc. 2015, 3, e00154-15. [Google Scholar] [CrossRef]
- Lindberg, A.A. Studies of a Receptor for Felix 0–1 Phage in Salmonella Minnesota. J. Gen. Microbiol. 1967, 48, 225–233. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lindberg, A.A.; Holme, T. Influence of O side chains on the attachment of the Felix O-1 bacteriophage to Salmonella bacteria. J. Bacteriol. 1969, 99, 513–519. [Google Scholar]
- Macphee, D.G.; Krishnapillai, V.; Roantree, R.J.; Stocker, B.A.D. Mutations in Salmonella typhimurium Conferring Resistance to Felix O Phage without Loss of Smooth Character. J. Gen. Microbiol. 1975, 87, 1–10. [Google Scholar] [CrossRef]
- Switt, A.I.M.; den Bakker, H.C.; Vongkamjan, K.; Hoelzer, K.; Warnick, L.D.; Cummings, K.J.; Wiedmann, M. Salmonella bacteriophage diversity reflects host diversity on dairy farms. Food Microbiol. 2013, 36, 275–285. [Google Scholar] [CrossRef]
- Akusobi, C.; Chan, B.K.; Williams, E.; Wertz, J.E.; Turner, P.E. Parallel evolution of host-attachment proteins in phage PP01 populations adapting to Escherichia coli O157:H7. Pharmaceuticals 2018, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.B.; Fernandes, E.; Carvalho, C.M.; Sillankorva, S.; Krylov, V.N.; Pleteneva, E.A.; Shaburova, O.V.; Nicolau, A.; Ferreira, E.C.; Azeredo, J. Selection and characterization of a multivalent Salmonella phage and its production in a nonpathogenic Escherichia coli strain. Appl. Environ. Microbiol. 2010, 76, 7338–7342. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Zhang, H.; Wang, R. Isolation and characterization of bacteriophages of Salmonella enterica serovar Pullorum. Poult. Sci. 2011, 90, 2370–2377. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.B.; Carvalho, C.; Azeredo, J.; Ferreira, E.C. Population dynamics of a Salmonella lytic phage and its host: Implications of the host bacterial growth rate in modelling. PLoS ONE 2014, 9, e102507. [Google Scholar] [CrossRef] [PubMed]
- Holguín, A.V.; Cárdenas, P.; Prada-Peñaranda, C.; Rabelo Leite, L.; Buitrago, C.; Clavijo, V.; Oliveira, G.; Leekitcharoenphon, P.; Møller Aarestrup, F.; Vives, M.J.; et al. Host Resistance, Genomics and Population Dynamics in a Salmonella Enteritidis and Phage System. Viruses 2019, 11, 188. [Google Scholar] [CrossRef]
- Wollin, R.; Creeger, E.S.; Rothfield, L.I.; Stocker, B.A.; Lindberg, A.A. Salmonella typhimurium mutants defective in UDP-d-galactose: Lipopolysaccharide α 1,6-d-galactosyltransferase. Structural, immunochemical, and enzymologic studies of rfaB mutants. J. Biol. Chem. 1983, 258, 3769–3774. [Google Scholar]
- Kaniuk, N.A.; Monteiro, M.A.; Parker, C.; Whitfield, C. Molecular diversity of the genetic loci responsible for lipopolysaccharide core oligosaccharide assembly within the genus Salmonella. Mol. Microbiol. 2002, 46, 1305–1318. [Google Scholar] [CrossRef]
- Morgan, E.; Campbell, J.D.; Rowe, S.C.; Bispham, J.; Stevens, M.P.; Bowen, A.J.; Barrow, P.A.; Maskell, D.J.; Wallis, T.S. Identification of host-specific colonization factors of Salmonella enterica serovar Typhimurium. Mol. Microbiol. 2004, 54, 994–1010. [Google Scholar] [CrossRef]
- Thompson , J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Geneious Prime version 2019.1.1. Available online: https://www.geneious.com (accessed on 3 March 2019).
- Hanada, K.; Shen, S.; Li, W. The Nonsynonymous/Synonymous Substitution Rate Ratio versus the Radical/Conservative Replacement Rate Ratio in the Evolution of Mammalian Genes. Mol. Biol. Evol. 2007, 24, 2235–2241. [Google Scholar] [CrossRef]
- Bartual, S.G.; Otero, J.M.; Garcia-Doval, C.; Llamas-Saiz, A.L.; Kahn, R.; Fox, G.C.; van Raaij, M.J. Structure of the bacteriophage T4 long tail fiber receptor-binding tip. Proc. Natl. Acad. Sci. USA 2010, 107, 20287–20292. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P.; van Raaij, M. Bacteriophage T4 long tail fiber domains. Biophys. Rev. 2018, 10, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Bartual, S.G.; Garcia-Doval, C.; Alonso, J.; Schoehn, G.; van Raaij, M.J. Two-chaperone assisted soluble expression and purification of the bacteriophage T4 long tail fibre protein gp37. Protein Expr. Purif. 2010, 70, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.C.T.; Friman, V.P.; Smith, M.C.M.; Brockhurst, M.A. Cross-resistance is modular in bacteria-phage interactions. PLoS Biol. 2018, 16, e2006057. [Google Scholar] [CrossRef] [PubMed]
- Grimont, P.A.; Weill, F.X. Antigenic formulae of the Salmonella serovars. In WHO Collaborating Centre for Reference and Research on Salmonella; Institut Pasteur: Paris, France, 2007; Volume 9, pp. 1–166. Available online: https://www.pasteur.fr/sites/default/files/veng_0.pdf (accessed on 3 March 2019).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteriophages | ||
|---|---|---|
| Characteristics | vB_Si_SF20-2 | vB_Si_QUI-1 |
| Host range classification | Wide host range (WHR) | Narrow host range (NHR) |
| Salmonella serovars lysed 1 | Choleraesuis, Panama, Javiana, Kentucky, Montevideo, Infantis, Oranienburg, Typhimurium, Corvallis, Mbandaka, Dublin, Newport, Braenderup, Enteritidis | Kentucky, Infantis, Oranienburg, Typhimurium, Dublin, Braenderup |
| NCBI Accession Number | MK965970.1 | MK965969.1 |
| BLAST results with highest e-value of fully annotated phage (accession) | Mushroom (KP143762.1) | Felix 01 (AF320576.1) |
| Genome size of assembly | 89,093 bp | 89,218 bp |
| G + C content | 39.76% | 39.14% |
| Size of the capsid 2 | 73 × 69 nm | 81 × 92 nm |
| Length of the tail | 144 ± 3 nm | 146 ± 3 nm |
| Burst size (viruses) | 30 ± 5 | 12.6 ± 4 |
| Latency time | 40 ± 10 min | 55 ± 15 min |
| Hours | Average 1 of Proportions of SI Survivors Exposed to WHR 2 Phage | Average of Proportions of SI Survivors Exposed to NHR 3 Phage |
|---|---|---|
| 1 | 8.1 × 10−6 | 8.8 × 10−6 |
| 2 | 4.2 × 10−6 | 1.0 × 10−5 |
| 3 | 8.8 × 10−6 | 4.0 × 10−5 |
| 4 | 8.8 × 10−7 | 3.1 × 10−6 |
| 5 | 3.3 × 10−7 | 2.0 × 10−6 |
| 6 | 2.1 × 10−6 | 7.7 × 10−6 |
| 7 | 3.5 × 10−6 | 1.2 × 10−5 |
| 8 | 7.6 × 10−7 | 1.5 × 10−6 |
| 9 | 8.5 × 10−7 | 1.2 × 10−6 |
| 10 | 1.2 × 10−6 | 1.8 × 10−6 |
| 11 | 8.1 × 10−6 | 1.2 × 10−6 |
| 12 | 4.2 × 10−6 | 9.7 × 10−7 |
| Average | 2.7 × 10−6 | 7.6 × 10−6 |
| Variant Information | SI Unexposed to WHR Phage (control) | SI Exposed to WHR Phage 12 h | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CDD and pfam ID 1 | Genome Position (contig) | Reference Allele | Alternative Allele | Kmer Coverage of Reference Allele | Kmer Coverage of Alternative Allele | Variant Frequency (%) | Kmer Coverage of Reference Allele | Kmer Coverage of Alternative Allele | Variant Frequency (%) | Protein Effect 7 |
| UDP-D-1,2 2 (PRK10124) | 94397 (4) | TG | T | 63 | 0 | 0.00 | 0 | 109 | 100.00 | Truncated protein 3 |
| Variant Information | SI unexposed to NHR phage (control) | SI exposed to NHR phage 12 Hour | ||||||||
| UDP-D-1,6 4 (PRK09922) | 92739 (4) | C | A | 92 | 0 | 0.00 | 0 | 64 | 100.00 | Radical Nonsynonymous Substitution |
| Variant Information of WHR | WHR phage (Control) | WHR phage 12 Hour | ||||||||
| Baseplate J-like protein (pfam04865) | 17509 (1) | G | T | 42 | 10 | 19.23 | 11 | 1 | 8.33 | Conservative Nonsynonymous Substitution |
| (FNI) 5_Phage protein (a) | 18756 (1) | C | A | 7 | 29 | 80.56 | 0 | 2 | 100.00 | Radical Nonsynonymous Substitution |
| (FNI) 5_Phage protein (b) | 18756 (1) | C | T | 7 | 24 | 77.42 | 0 | 3 | 100.00 | Conservative Nonsynonymous Substitution |
| gp37-like 6 (pfam12604) | 21980 (1) | C | A | 27 | 30 | 52.63 | 0 | 5 | 100.00 | Radical Nonsynonymous Substitution |
| gp37-like 6 (pfam12604) | 22092 (1) | T | C | 21 | 32 | 60.38 | 0 | 6 | 100.00 | Conservative Nonsynonymous Substitution |
| Variant Information of NHR | NHR phage (Control) | NHR phage 12 Hour | ||||||||
| Baseplate J-like protein (pfam04865) | 17384 (1) | G | A | 7 | 8 | 53.33 | 5 | 17 | 77.27 | Radical Nonsynonymous Substitution |
| (NI)_Hypothetical protein | 40239 (1) | T | G | 3 | 1 | 25.00 | 13 | 0 | 0.00 | Radical Nonsynonymous Substitution |
| S. Infantis Obtained from Exposure to Wide-Host-Range (WHR) Phage | S. Infantis Obtained from Exposure to Narrow-Host-Range (NHR) Phage | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EOP 1 of Each Replicate (Replicates) | EOP 1 of Each Replicate (Replicates) | |||||||||
| Phage | (R1) | (R2) | (R3) | (R4) | Average of replicates | (R1) | (R2) | (R3) | (R4) | Average of replicates |
| Wild-type WHR phage | 0.2 | 1 | 0 | 0.1 | 0.3 | 0 | 0 | 0 | 0 | 0 |
| Wild-type NHR phage | 1 | 0.1 | 0 | 1 | 0.5 | 0 | 0 | 0 | 0 | 0 |
| Host range assay of wide-host-range phage obtained from each replicate (Nº of a given replicate) | Host range of narrow-host-range phage obtained from each replicate (Nº of a given replicate) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Wild-type | R1 | R2 | R3 | R4 | Wild-type | R1 | R2 | R3 | R4 | |
| Serovars lysed * | CHO PAN JAV KEN MON INF ** ORA TYP COR MBA DUB NEW BRA ENT | MUE VIR SAI CHO PAN JAV KEN MON INF ** ORA TYP AGO HEI NEW STA 4,5,12:i:- | VIR, SAI CHO PAN JAV MON INF ** ORA TYP AGO HEI NEW 4,5,12:i:- | CHO MON INF ** ORA | CHO MON INF ** BRA | KEN INF ** ORA TYP DUB BRA | ORA NEW | ORA TYP PAN COR NEW | VIR JAV ORA NEW | ORA NEW |
| Total serovars lysed | 14 | 16 | 12 | 4 | 4 | 6 | 2 | 5 | 4 | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivera, D.; Hudson, L.K.; Denes, T.G.; Hamilton-West, C.; Pezoa, D.; Moreno-Switt, A.I. Two Phages of the Genera Felixunavirus Subjected to 12 Hour Challenge on Salmonella Infantis Showed Distinct Genotypic and Phenotypic Changes. Viruses 2019, 11, 586. https://doi.org/10.3390/v11070586
Rivera D, Hudson LK, Denes TG, Hamilton-West C, Pezoa D, Moreno-Switt AI. Two Phages of the Genera Felixunavirus Subjected to 12 Hour Challenge on Salmonella Infantis Showed Distinct Genotypic and Phenotypic Changes. Viruses. 2019; 11(7):586. https://doi.org/10.3390/v11070586
Chicago/Turabian StyleRivera, Dácil, Lauren K. Hudson, Thomas G. Denes, Christopher Hamilton-West, David Pezoa, and Andrea I. Moreno-Switt. 2019. "Two Phages of the Genera Felixunavirus Subjected to 12 Hour Challenge on Salmonella Infantis Showed Distinct Genotypic and Phenotypic Changes" Viruses 11, no. 7: 586. https://doi.org/10.3390/v11070586
APA StyleRivera, D., Hudson, L. K., Denes, T. G., Hamilton-West, C., Pezoa, D., & Moreno-Switt, A. I. (2019). Two Phages of the Genera Felixunavirus Subjected to 12 Hour Challenge on Salmonella Infantis Showed Distinct Genotypic and Phenotypic Changes. Viruses, 11(7), 586. https://doi.org/10.3390/v11070586