Avirulins, a Novel Class of HIV-1 Reverse Transcriptase Inhibitors Effective in the Female Reproductive Tract Mucosa

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Viruses
2.2. Collection of Human Specimens
2.3. TZM-bl Luciferase Infection Assay
2.4. In Silico Compound Screening
2.5. PM1 and PBMC Infection and p24
2.6. Cytotoxicity and Cell Viability
2.7. Reverse Transcriptase Inhibition Assay
2.8. Avirulin Antiviral Activity in Clarified Vaginal Fluid
3. Results
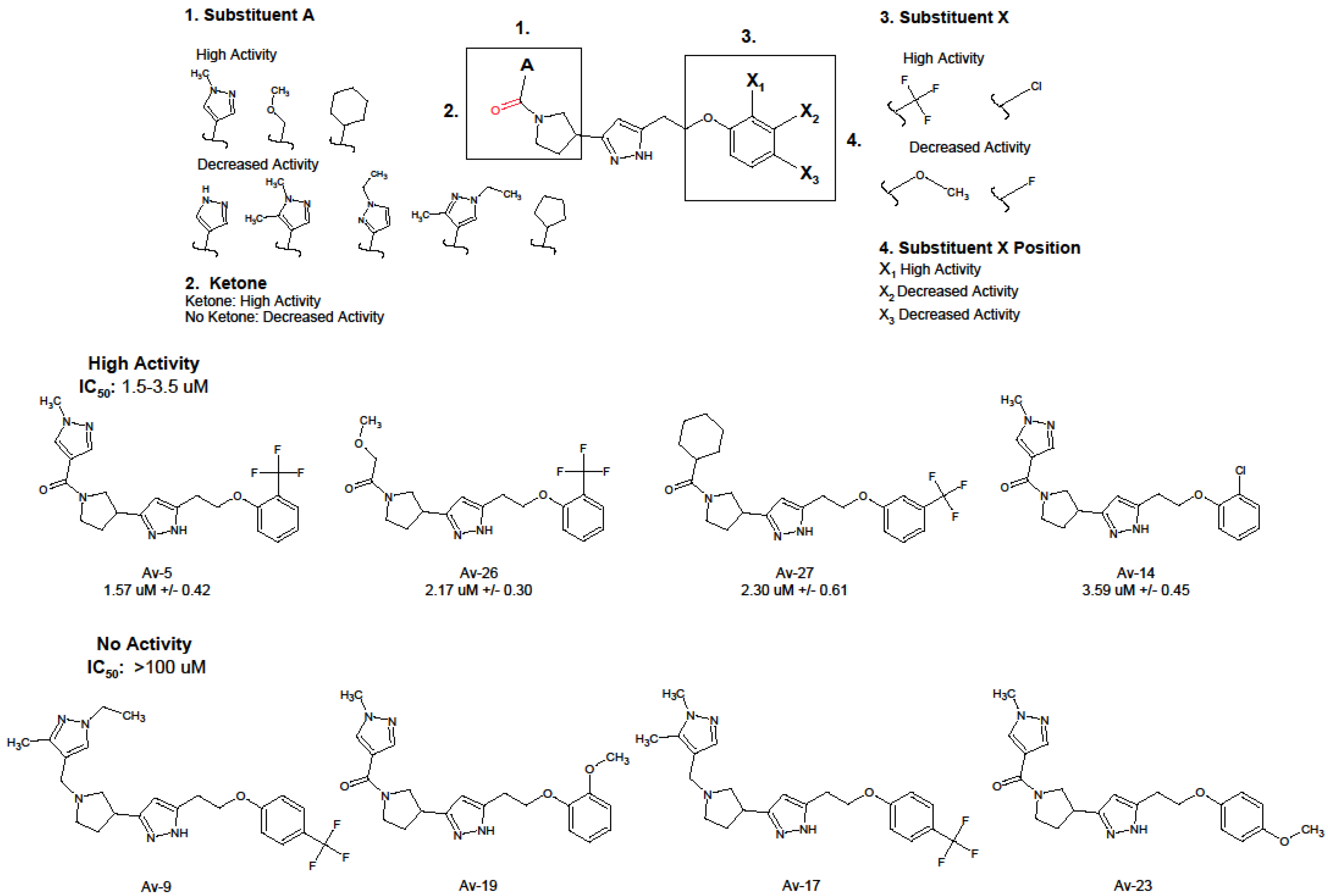
3.1. Antiviral and Structural Characteristics of Avirulins
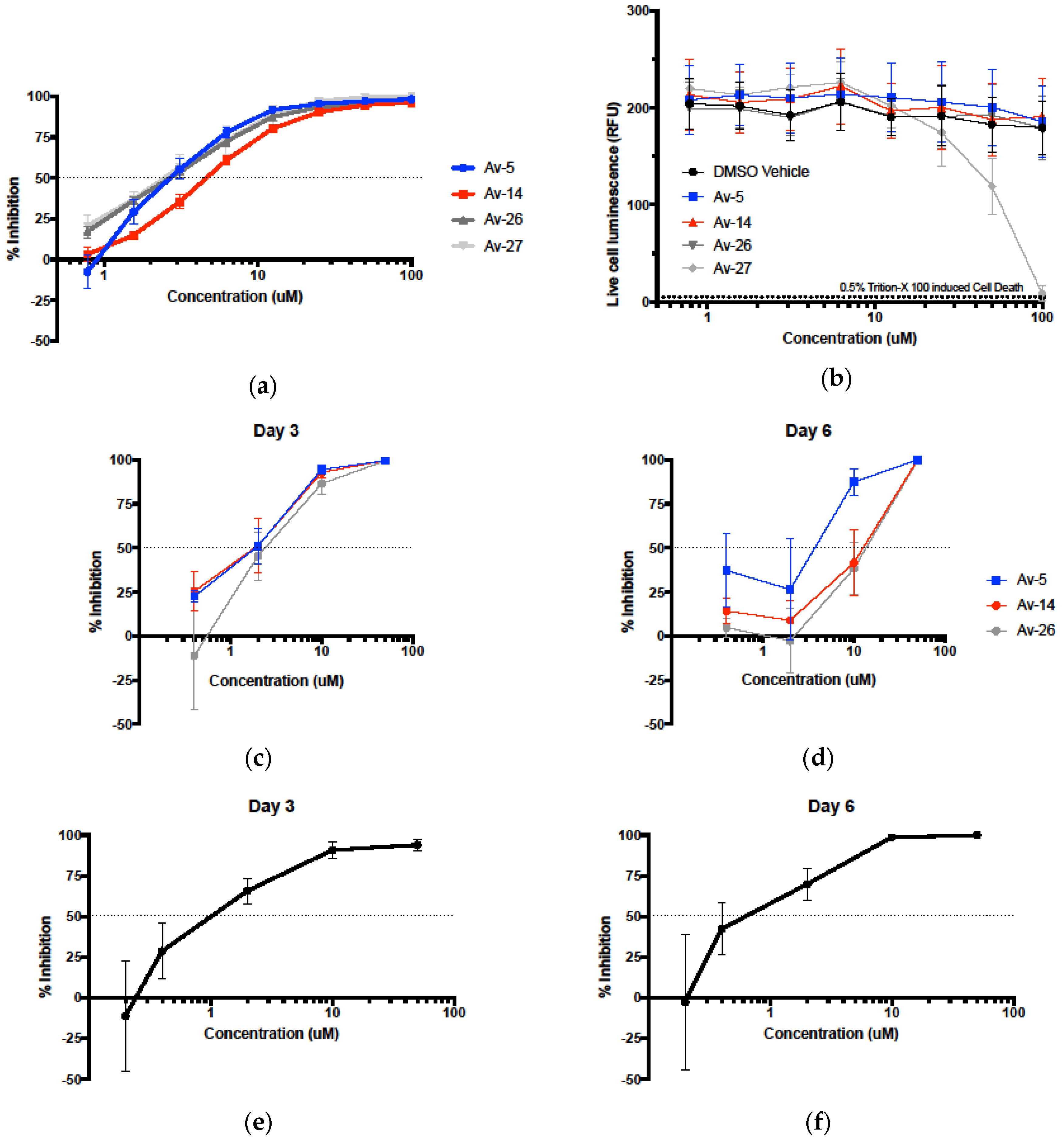
3.2. Evaluation of Antiviral Activity in Lead Compounds
3.3. HIV-1 Reverse Transcriptase Inhibition of Avirulins
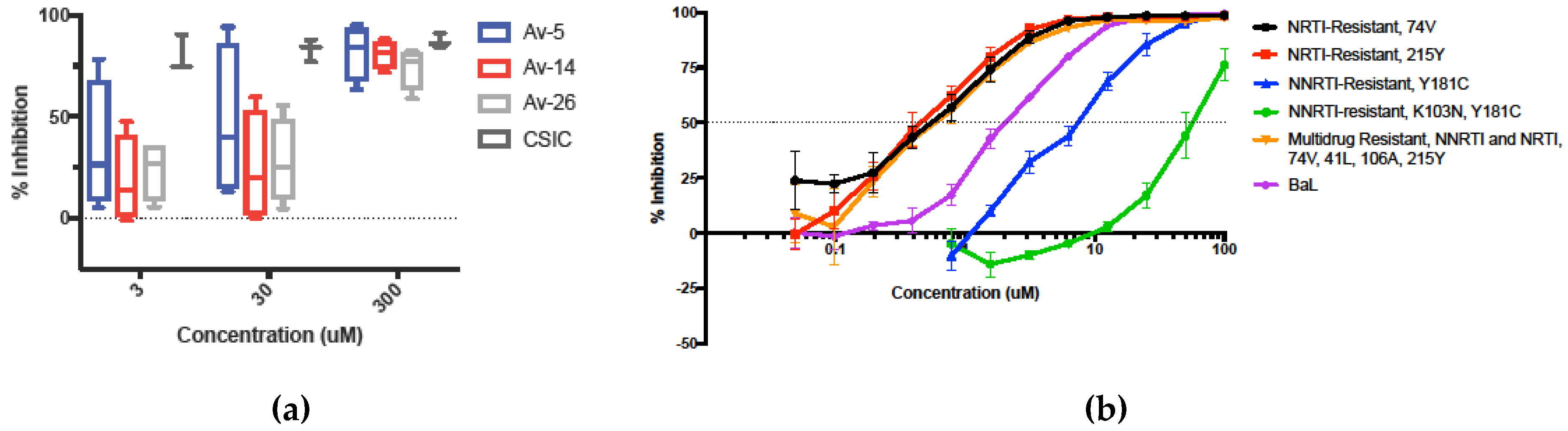
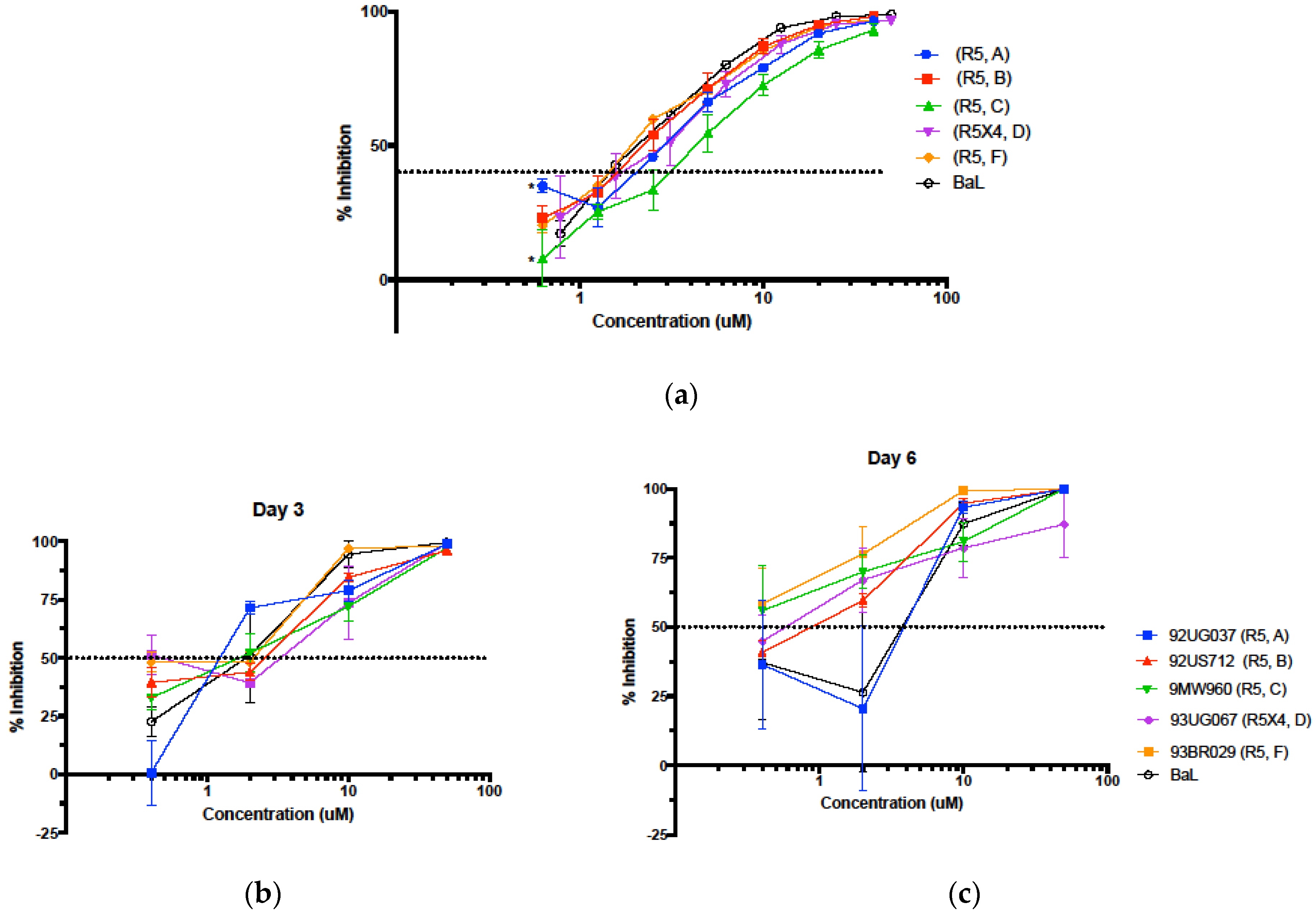
3.4. Inhibition in Genetically Variant HIV-1 Clades HIV
3.5. Evaluation in the Female Reproductive Tract
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gallo, R.C.; Salahuddin, S.Z.; Popovic, M.; Shearer, G.M.; Kaplan, M.; Haynes, B.F.; Palker, T.J.; Redfield, R.; Oleske, J.; Safai, B.; et al. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 1984, 224, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.A. Virus-host interactions in HIV pathogenesis: Directions for therapy. Adv. Dent. Res. 2011, 23, 13–18. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Veazey, R.S.; Lackner, A.A. HIV swiftly guts the immune system. Nat. Med. 2005, 11, 469–470. [Google Scholar] [CrossRef]
- Pantaleo, G.; Fauci, A.S. Immunopathogenesis of HIV infection. Annu. Rev. Microbiol. 1996, 50, 825–854. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deeks, S.G.; Lewin, S.R.; Havlir, D.V. The end of AIDS: HIV infection as a chronic disease. Lancet 2013, 382, 1525–1533. [Google Scholar] [CrossRef]
- UN Joint Programme on HIV/AIDS (UNAIDS), Global AIDS Update—2018. Available online: http://www.unaids.org/en/20180718_GR2018 (accessed on 1 April 2019).
- Wheeler, W.H.; Ziebell, R.A.; Zabina, H.; Pieniazek, D.; Prejean, J.; Bodnar, U.R.; Mahle, K.C.; Heneine, W.; Johnson, J.A.; Hall, H.I.; et al. Prevalence of transmitted drug resistance associated mutations and HIV-1 subtypes in new HIV-1 diagnoses, U.S.–2006. AIDS 2010, 24, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Dahabieh, M.S.; Battivelli, E.; Verdin, E. Understanding HIV Latency: The Road to an HIV Cure. Annu. Rev. Med. 2015, 66, 407–421. [Google Scholar] [CrossRef]
- Richman, D.D.; Margolis, D.M.; Delaney, M.; Greene, W.C.; Hazuda, D.; Pomerantz, R.J. The Challenge of Finding a Cure for HIV Infection. Science 2009, 323, 1304–1307. [Google Scholar] [CrossRef]
- Mascola, J.R. The modern era of HIV-1 vaccine development. Science 2015, 349, 139–140. [Google Scholar] [CrossRef]
- Haynes, B.F.; Burton, D.R. Developing an HIV vaccine. Science 2017, 355, 1129–1130. [Google Scholar] [CrossRef]
- Spinner, C.D.; Boesecke, C.; Zink, A.; Jessen, H.; Stellbrink, H.-J.; Rockstroh, J.K.; Esser, S. HIV pre-exposure prophylaxis (PrEP): A review of current knowledge of oral systemic HIV PrEP in humans. Infection 2016, 44, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Shattock, R.J.; Moore, J.P. Inhibiting sexual transmission of HIV-1 infection. Nat. Rev. Microbiol. 2003, 1, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Dayal, M.B.; Wheeler, J.; Williams, C.J.; Barnhart, K.T. Disruption of the upper female reproductive tract epithelium by nonoxynol-9. Contraception 2003, 68, 273–279. [Google Scholar] [CrossRef]
- Smith, D.A.; Di, L.; Kerns, E.H. The effect of plasma protein binding on in vivo efficacy: Misconceptions in drug discovery. Nat. Rev. Drug. Discov. 2010, 9, 929–939. [Google Scholar] [CrossRef]
- Marie, B.; Mørck, N.H. Mucus as a Barrier to Drug Delivery – Understanding and Mimicking the Barrier Properties. Basic Clin. Pharmacol. Toxicol. 2015, 116, 179–186. [Google Scholar]
- Manallack, D.T.; Prankerd, R.J.; Yuriev, E.; Oprea, T.I.; Chalmers, D.K. The Significance of Acid/Base Properties in Drug Discovery. Chem. Soc. Rev. 2013, 42, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Larder, B.A.; Kellam, P.; Kemp, S.D. Convergent combination therapy can select viable multidrug-resistant HIV-1 in vitro. Nature 1993, 365, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Richman, D.D.; Havlir, D.; Corbeil, J.; Looney, D.; Ignacio, C.; Spector, S.A.; Sullivan, J.; Cheeseman, S.; Barringer, K.; Pauletti, D. Nevirapine resistance mutations of human immunodeficiency virus type 1 selected during therapy. J. Virol. 1994, 68, 1660–1666. [Google Scholar]
- Esté, J.A.; Cihlar, T. Current status and challenges of antiretroviral research and therapy. Antiviral Res. 2010, 85, 25–33. [Google Scholar] [CrossRef]
- Hu, W.S.; Hughes, S.H. HIV-1 Reverse Transcription. Cold Spring Harb. Perspect. Med. 2012, 2, a006882. [Google Scholar] [CrossRef]
- Craigie, R.; Bushman, F.D. HIV DNA Integration. Cold Spring Harb. Perspect. Med. 2012, 2, a006890. [Google Scholar] [CrossRef]
- Brik, A.; Wong, C.H. HIV-1 protease: Mechanism and drug discovery. Org. Biomol. Chem. 2003, 1, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; et al. Structure of the CCR5 Chemokine Receptor–HIV Entry Inhibitor Maraviroc Complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Pau, A.K.; George, J.M. Antiretroviral Therapy. Infect. Dis. Clin. N. Am. 2014, 28, 371–402. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.J.; Beusenberg, M.; Habiyambere, V.; Shaffer, N.; Vitoria, M.A.; Montero, R.G.; Easterbrook, P.J.; Doherty, M.C. Adoption of national recommendations related to use of antiretroviral therapy before and shortly following the launch of the 2013 WHO consolidated guidelines. AIDS 2014, 28, S217–S224. [Google Scholar] [CrossRef] [PubMed]
- Thigpen, M.C.; Kebaabetswe, P.M.; Paxton, L.A.; Smith, D.K.; Rose, C.E.; Segolodi, T.M.; Henderson, F.L.; Pathak, S.R.; Soud, F.A.; Chillag, K.L.; et al. Antiretroviral preexposure prophylaxis for heterosexual HIV transmission in Botswana. N. Engl. J. Med. 2012, 367, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Platt, E.J.; Bilska, M.; Kozak, S.L.; Kabat, D.; Montefiori, D.C. Evidence that Ecotropic Murine Leukemia Virus Contamination in TZM-bl Cells Does Not Affect the Outcome of Neutralizing Antibody Assays with Human Immunodeficiency Virus Type 1. J. Virol. 2009, 83, 8289–8292. [Google Scholar] [CrossRef] [PubMed]
- Lusso, P.; Cocchi, F.; Balotta, C.; Markham, P.D.; Louie, A.; Farci, P.; Pal, R.; Gallo, R.C.; Reitz, M.S., Jr. Growth of macrophage-tropic and primary human immunodeficiency virus type 1 (HIV-1) isolates in a unique CD4+ T-cell clone (PM1): Failure to downregulate CD4 and to interfere with cell-line-tropic HIV-1. J. Virol. 1995, 69, 3712–3720. [Google Scholar]
- St Clair, M.H.; Martin, J.L.; Tudor-Williams, G.; Bach, M.C.; Vavro, C.L.; King, D.M.; Kellam, P.; Kemp, S.D.; Larder, B.A. Resistance to ddI and sensitivity to AZT induced by a mutation in HIV-1 reverse transcriptase. Science 1991, 253, 1557–1559. [Google Scholar] [CrossRef]
- Williams, T.M.; Ciccarone, T.M.; MacTough, S.C.; Rooney, C.S.; Balani, S.K.; Condra, J.H.; Emini, E.A.; Goldman, M.E.; Greenlee, W.J. 5-Chloro-3-(phenylsulfonyl)indole-2-carboxamide: A novel, non-nucleoside inhibitor of HIV-1 reverse transcriptase. J. Med. Chem. 1993, 36, 1291–1294. [Google Scholar] [CrossRef]
- Wagner, A.B. SciFinder Scholar 2006: An Empirical Analysis of Research Topic Query Processing. J. Chem. Inf. Model. 2006, 46, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Cherne, M.D.; Hall, J.; Kellner, A.; Chong, C.F.; Cole, A.L.; Cole, A.M. Assessment of Avirulin HIV-1 Protease and Integrase Inhibition; University of Central Florida: Orlando, FL, USA, 2017. [Google Scholar]
- Preston, B.D.; Dougherty, J.P. Mechanisms of retroviral mutation. Trends Microbiol. 1996, 4, 16–21. [Google Scholar] [CrossRef]
- Shattock, R.J.; Rosenberg, Z. Microbicides: Topical Prevention against HIV. Cold Spring Harb. Perspect. Med. 2012, 2, a007385. [Google Scholar] [CrossRef] [PubMed]
- Fichorova, R.N.; Anderson, D.J. Differential expression of immunobiological mediators by immortalized human cervical and vaginal epithelial cells. Biol. Reprod. 1999, 60, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Shi, S. Biologics: An update and challenge of their pharmacokinetics. Curr. Drug Metabol. 2014, 15, 271–290. [Google Scholar] [CrossRef]
- Ghosh, M.; Fahey, J.V.; Shen, Z.; Lahey, T.; Cu-Uvin, S.; Wu, Z.; Mayer, K.; Wright, P.F.; Kappes, J.C.; Ochsenbauer, C.; et al. Anti-HIV activity in cervical-vaginal secretions from HIV-positive and -negative women correlate with innate antimicrobial levels and IgG antibodies. PLoS ONE 2010, 5, e11366. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.L.; Ghosh, M.; Raker, C.; Fahey, J.; Song, Y.; Rouse, D.J.; Wira, C.R.; Cu-Uvin, S. In vitro anti-HIV-1 activity in cervicovaginal secretions from pregnant and nonpregnant women. Am. J. Obstet. Gynecol. 2012, 207, 65.e1–65.e10. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.V.; Ghosh, M.; Fahey, J.V.; Ochsenbauer, C.; Rossoll, R.M.; Wira, C.R. Innate immunity in the vagina (Part II): Anti-HIV activity and antiviral content of human vaginal secretions. Am. J. Reprod. Immunol. 2014, 72, 22–33. [Google Scholar] [CrossRef]
- Venier, O.; Pascal, C.; Braun, A.; Namane, C.; Mougenot, P.; Crespin, O.; Pacquet, F.; Mougenot, C.; Monseau, C.; Onofri, B.; et al. Pyrrolidine-pyrazole ureas as potent and selective inhibitors of 11β-hydroxysteroid-dehydrogenase type 1. Bioorg. Med. Chem. Lett. 2011, 21, 2244–2251. [Google Scholar] [CrossRef]
- Luber, A.D. Genetic barriers to resistance and impact on clinical response. MedGenMed 2005, 7, 69. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Gonzales, M.J.; Kantor, R.; Betts, B.J.; Ravela, J.; Shafer, R.W. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003, 31, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J.; Perry, C.M. Delavirdine. Drugs 2000, 60, 1411–1444. [Google Scholar] [CrossRef] [PubMed]
- Kantor, R.; Katzenstein, D. Drug resistance in non-subtype B HIV-1. J. Clin. Virol. 2004, 29, 152–159. [Google Scholar] [CrossRef]
- Spira, S.; Wainberg, M.A.; Loemba, H.; Turner, D.; Brenner, B.G. Impact of clade diversity on HIV-1 virulence, antiretroviral drug sensitivity and drug resistance. J. Antimicrob. Chemother. 2003, 51, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Tornesello, M.L.; Buonaguro, F.M. Human Immunodeficiency Virus Type 1 Subtype Distribution in the Worldwide Epidemic: Pathogenetic and Therapeutic Implications. J. Virol. 2007, 81, 10209–10219. [Google Scholar] [CrossRef] [PubMed]
- Loemba, H.; Brenner, B.; Parniak, M.A.; Ma’ayan, S.; Spira, B.; Moisi, D.; Oliveira, M.; Detorio, M.; Essex, M.; Wainberg, M.A. Polymorphisms of cytotoxic T-lymphocyte (CTL) and T-helper epitopes within reverse transcriptase (RT) of HIV-1 subtype C from Ethiopia and Botswana following selection of antiretroviral drug resistance. Antivir. Res. 2002, 56, 129–142. [Google Scholar] [CrossRef]
- Sutherland, K.A.; Ghosn, J.; Gregson, J.; Mbisa, J.L.; Chaix, M.L.; Cohen Codar, I.; Delfraissy, J.F.; Delaugerre, C.; Gupta, R.K. HIV-1 subtype influences susceptibility and response to monotherapy with the protease inhibitor lopinavir/ritonavir. J. Antimicrob. Chemother. 2015, 70, 243–248. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pendse, R.; Gupta, S.; Yu, D.; Sarkar, S. HIV/AIDS in the South-East Asia region: Progress and challenges. J. Virus Erad. 2016, 2, 1–6. [Google Scholar] [PubMed]
- Das, K.; Arnold, E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr. Opin. Virol. 2013, 3, 111–118. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IUPAC | IC50 (uM) 1 | IC90 (uM) 1 |
|---|---|---|---|
| Av-5 | 3-[1-(1-methyl-1H-pyrazole-4-carbonyl)pyrrolidin-3-yl]-5-{2-[2-(trifluoromethyl)phenoxy]ethyl}-1H-pyrazole | 1.57 ± 0.42 | 8.41 ± 1.26 |
| Av-26 | 2-methoxy-1-[3-(5-{2-[2-(trifluoromethyl)phenoxy]ethyl}-1H-pyrazol-3-yl)pyrrolidin-1-yl]ethan-1-one | 2.17 ± 0.30 | 15.9 ± 1.21 |
| Av-27 | 3-(1-cyclohexanecarbonylpyrrolidin-3-yl)-5-{2-[3-(trifluoromethyl)phenoxy]ethyl}-1H-pyrazole | 2.30 ± 0.61 | 18.2 ± 1.61 |
| Av-14 | 5-[2-(2-chlorophenoxy)ethyl]-3-[1-(1-methyl-1H-pyrazole-4-carbonyl)pyrrolidin-3-yl]-1H-pyrazole | 3.59 ± 0.45 | 18.65 ± 1.18 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cherne, M.D.; Hall, J.; Kellner, A.; Chong, C.F.; Cole, A.L.; Cole, A.M. Avirulins, a Novel Class of HIV-1 Reverse Transcriptase Inhibitors Effective in the Female Reproductive Tract Mucosa. Viruses 2019, 11, 408. https://doi.org/10.3390/v11050408
Cherne MD, Hall J, Kellner A, Chong CF, Cole AL, Cole AM. Avirulins, a Novel Class of HIV-1 Reverse Transcriptase Inhibitors Effective in the Female Reproductive Tract Mucosa. Viruses. 2019; 11(5):408. https://doi.org/10.3390/v11050408
Chicago/Turabian StyleCherne, Michelle D., Jesse Hall, Alisha Kellner, Christine F. Chong, Amy L. Cole, and Alexander M. Cole. 2019. "Avirulins, a Novel Class of HIV-1 Reverse Transcriptase Inhibitors Effective in the Female Reproductive Tract Mucosa" Viruses 11, no. 5: 408. https://doi.org/10.3390/v11050408
APA StyleCherne, M. D., Hall, J., Kellner, A., Chong, C. F., Cole, A. L., & Cole, A. M. (2019). Avirulins, a Novel Class of HIV-1 Reverse Transcriptase Inhibitors Effective in the Female Reproductive Tract Mucosa. Viruses, 11(5), 408. https://doi.org/10.3390/v11050408