Coxsackievirus B3 Responds to Polyamine Depletion via Enhancement of 2A and 3C Protease Activity


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
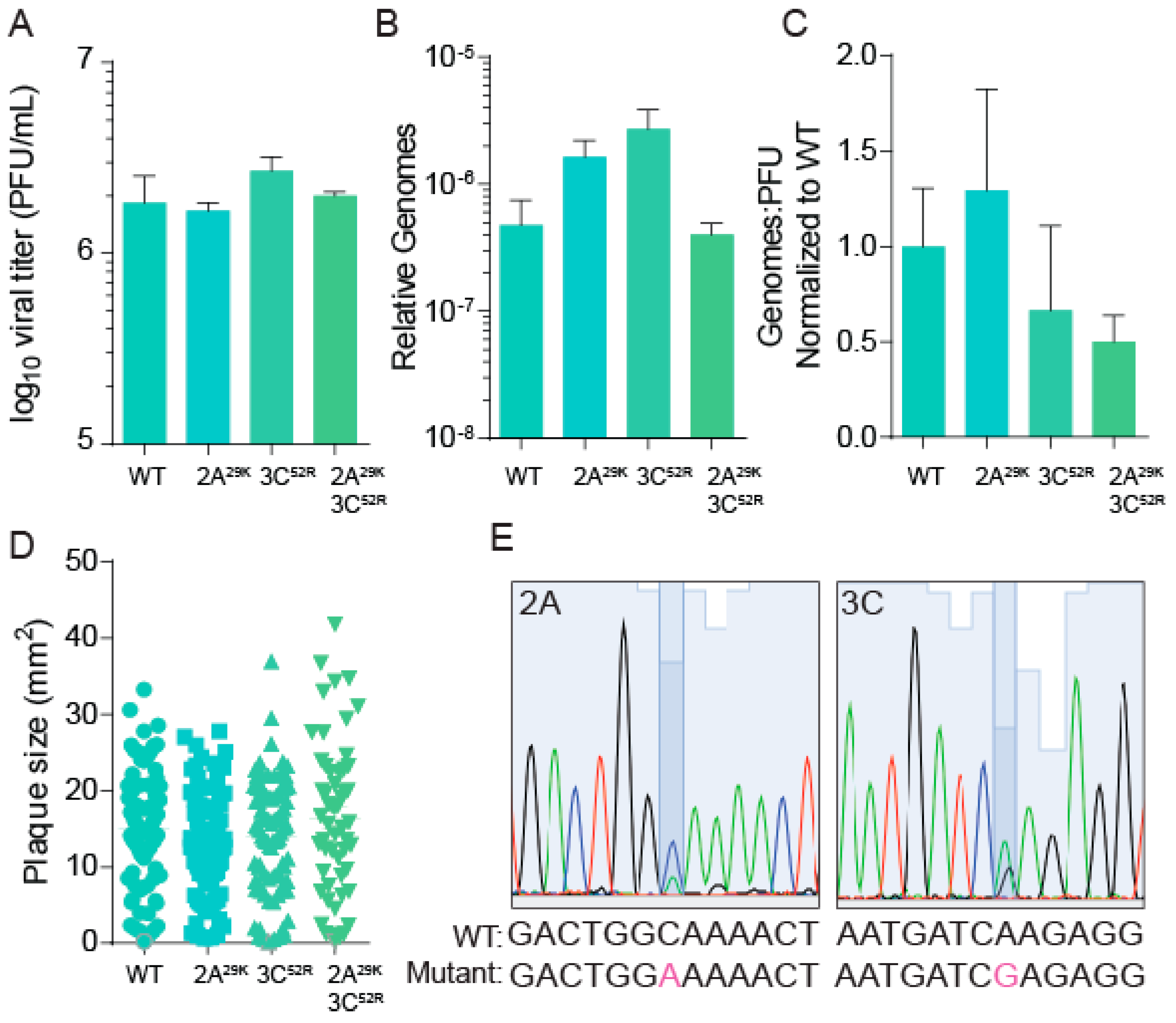
2.2. Generation of CVB3 and 2A29K and 3C52R Mutants
2.3. Infection and Enumeration of Viral Titers
2.4. Mutant CVB3 Propagation
2.5. Drug Treatments
2.6. RNA Purification and cDNA Synthesis
2.7. DFMO and DENSpm Sensitivity Assays
2.8. Stability and Fitness Assays
2.9. Protease Plasmid Cloning
2.10. Transfections
2.11. Luciferase Protease Assay
2.12. Western Blot
2.13. Plaque Size Measurement
2.14. Thin Layer Chromatography Determination of Polyamines
2.15. Statistical Analysis
3. Results
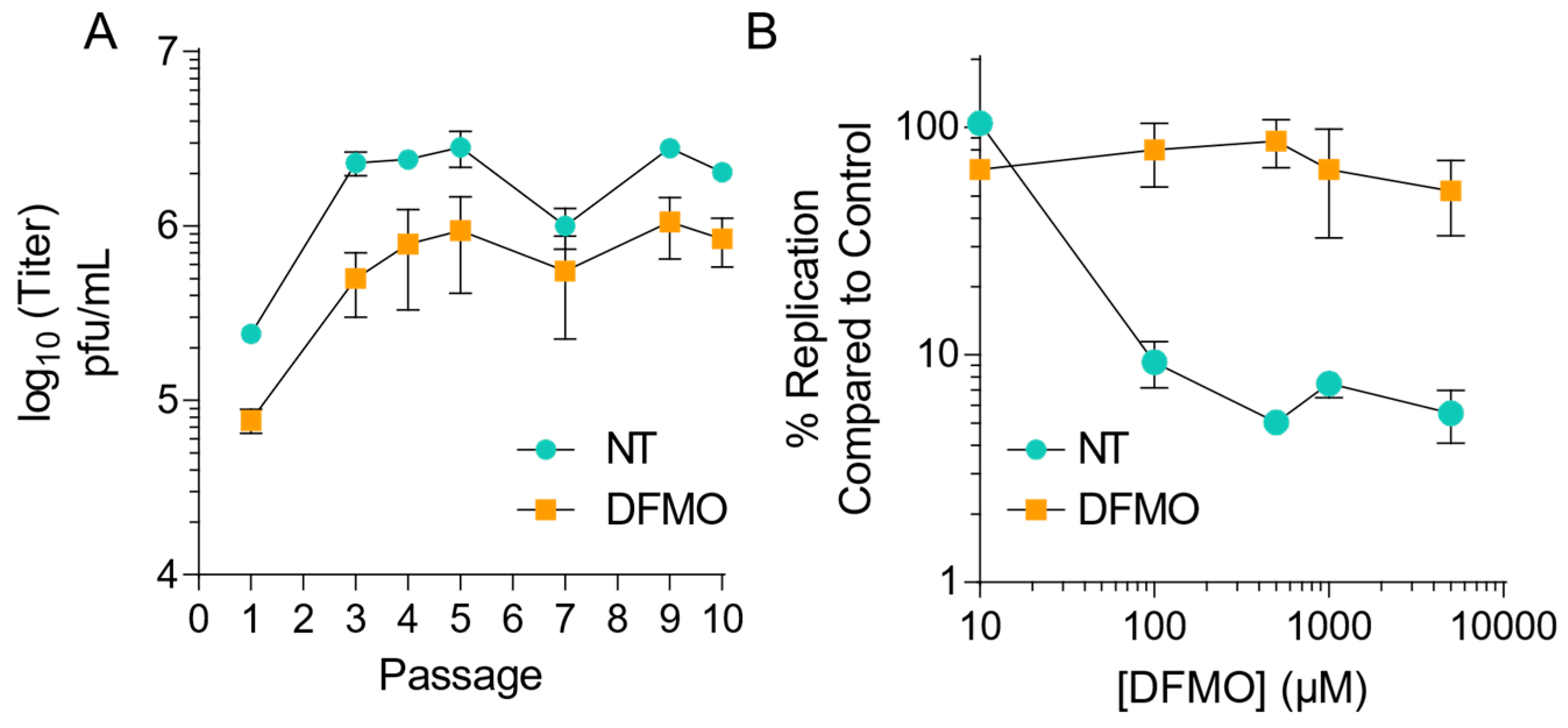
3.1. CVB3 Gains Resistance to Polyamine Depletion over Passages with DFMO
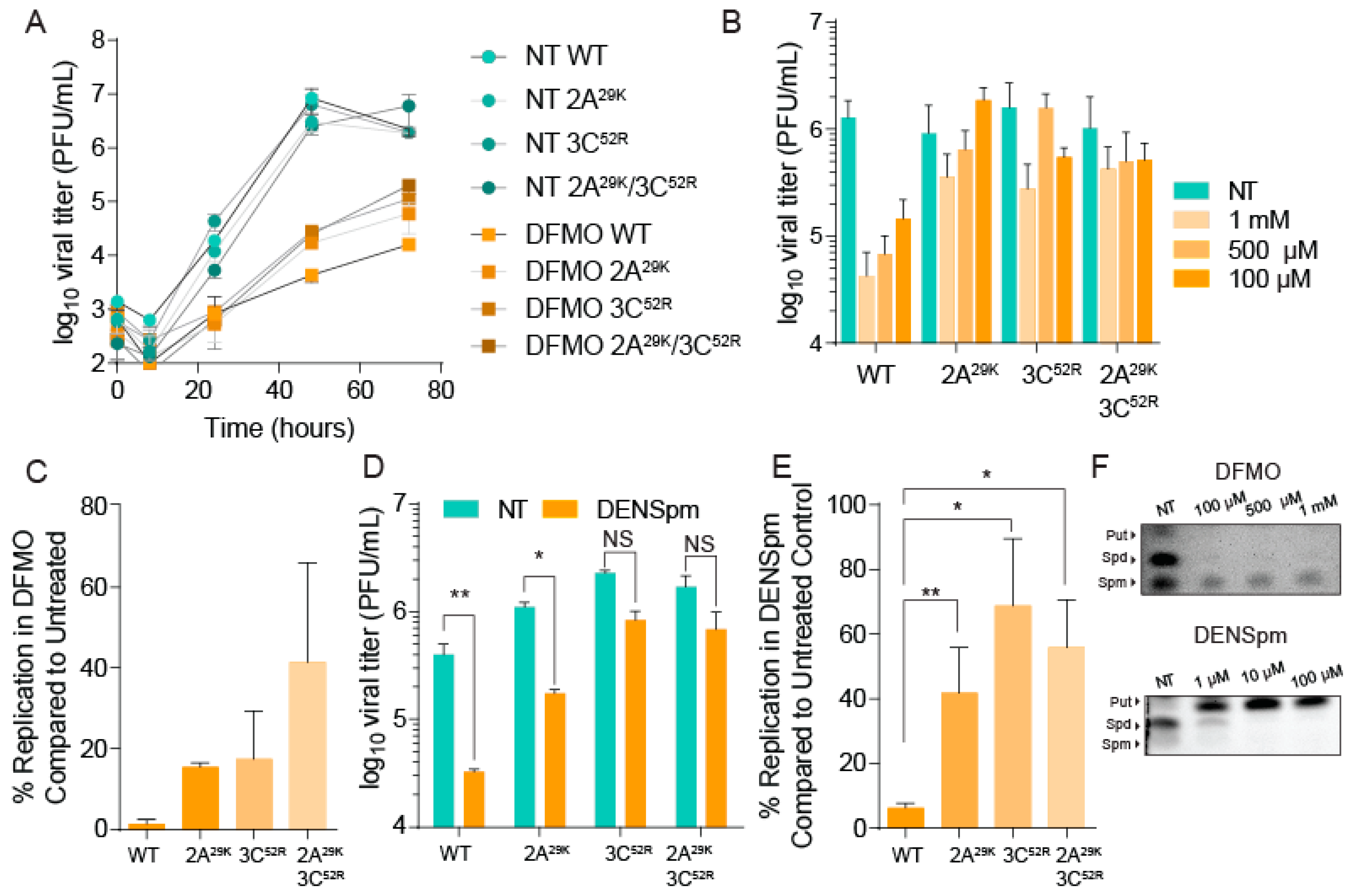
3.2. CVB3 Mutations in 2A and 3C Proteases Confer Resistance to Polyamine Depletion
3.3. CVB3 Stability, Fitness, and Specific Infectivity Are Not Altered with Protease Mutation
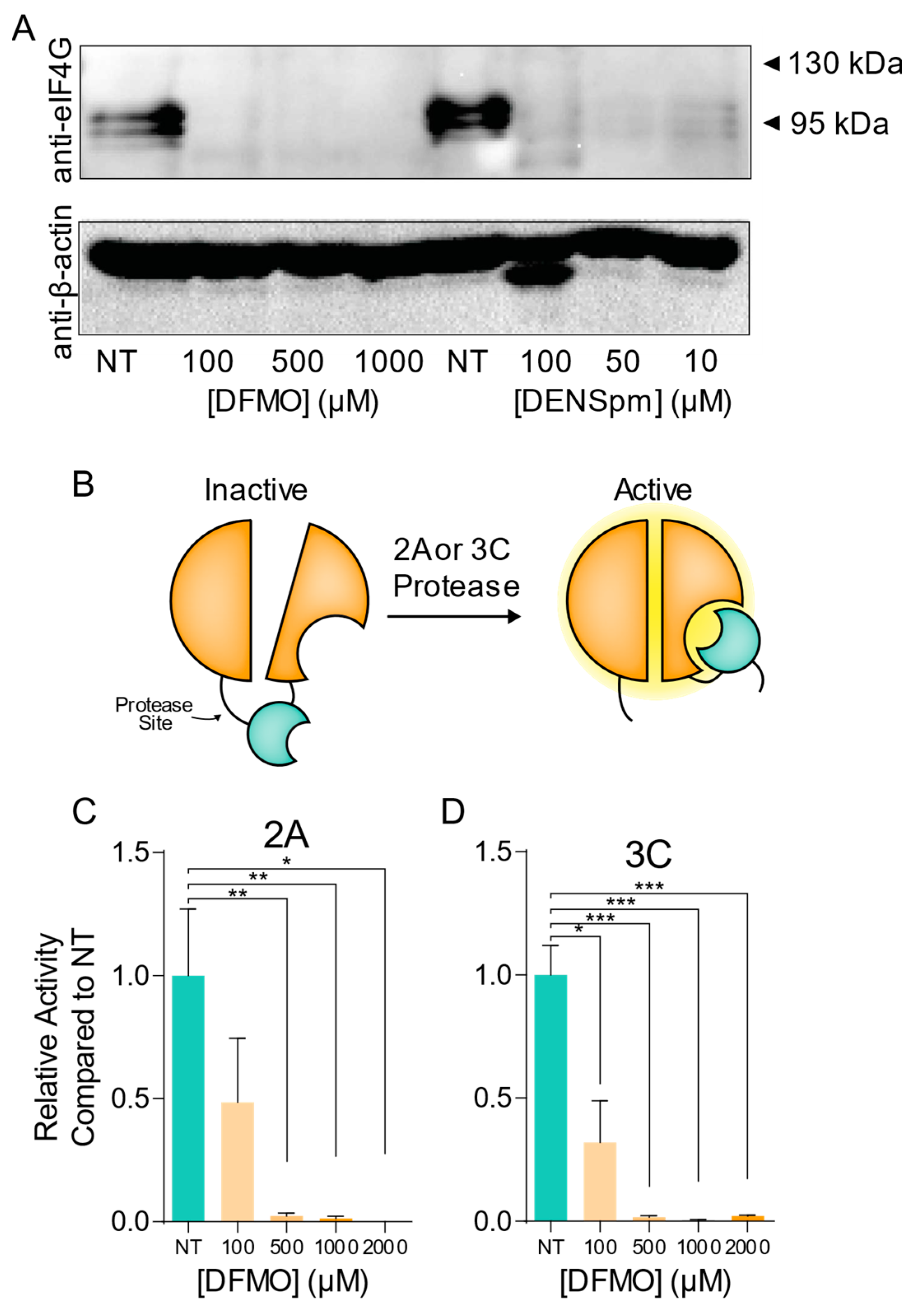
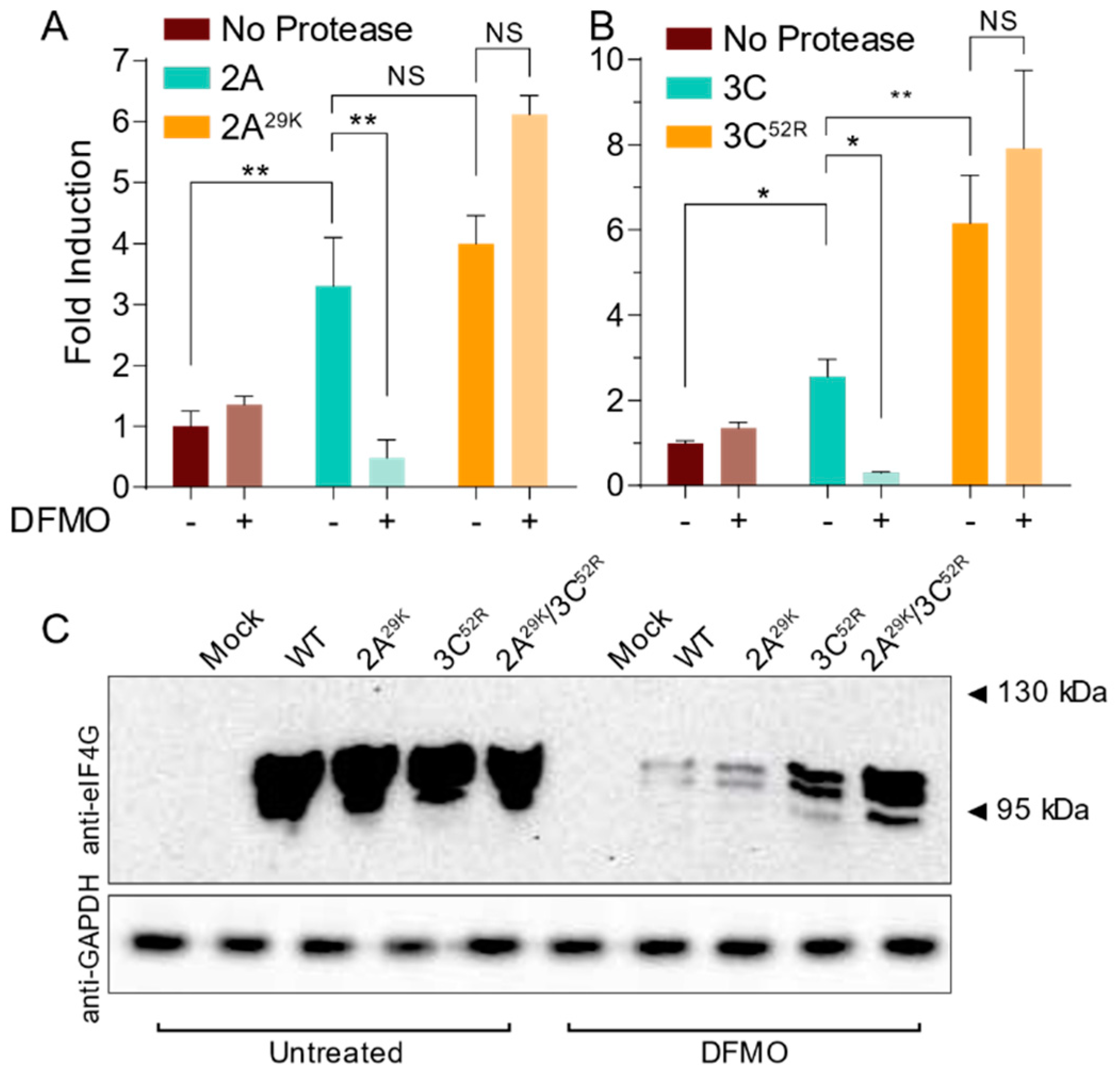
3.4. CVB3 Protease Activity Is Modulated by Cellular Polyamine Levels
3.5. CVB3 Protease Mutations Enhance Proteolytic Activity in Polyamine-Depleted Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chapman, N.M.; Kim, K.-S. Persistent Coxsackievirus Infection: Enterovirus Persistence in Chronic Myocarditis and Dilated Cardiomyopathy. In Group B Coxsackieviruses; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2008; pp. 275–292. ISBN 978-3-540-75545-6. [Google Scholar]
- Matteucci, D.; Paglianti, M.; Giangregorio, A.M.; Capobianchi, M.R.; Dianzani, F.; Bendinelli, M. Group B coxsackieviruses readily establish persistent infections in human lymphoid cell lines. J. Virol. 1985, 56, 651–654. [Google Scholar]
- Laitinen, O.H.; Svedin, E.; Kapell, S.; Nurminen, A.; Hytönen, V.P.; Flodström-Tullberg, M. Enteroviral proteases: Structure, host interactions and pathogenicity. Rev. Med. Virol. 2016, 26, 251–267. [Google Scholar] [CrossRef]
- Lyoo, H.; Dorobantu, C.M.; van der Schaar, H.M.; van Kuppeveld, F.J.M. Modulation of proteolytic polyprotein processing by coxsackievirus mutants resistant to inhibitors targeting phosphatidylinositol-4-kinase IIIβ or oxysterol binding protein. Antiviral Res. 2017, 147, 86–90. [Google Scholar] [CrossRef]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef]
- Glaser, W.; Skern, T. Extremely efficient cleavage of eIF4G by picornaviral proteinases L and 2A in vitro. FEBS Lett. 2000, 480, 151–155. [Google Scholar] [CrossRef]
- Wu, S.; Wang, Y.; Lin, L.; Si, X.; Wang, T.; Zhong, X.; Tong, L.; Luan, Y.; Chen, Y.; Li, X.; et al. Protease 2A induces stress granule formation during coxsackievirus B3 and enterovirus 71 infections. Virol. J. 2014, 11, 192. [Google Scholar] [CrossRef]
- Hanson, P.J.; Ye, X.; Qiu, Y.; Zhang, H.M.; Hemida, M.G.; Wang, F.; Lim, T.; Gu, A.; Cho, B.; Kim, H.; et al. Cleavage of DAP5 by coxsackievirus B3 2A protease facilitates viral replication and enhances apoptosis by altering translation of IRES-containing genes. Cell Death Differ. 2016, 23, 828–840. [Google Scholar] [CrossRef]
- Qiu, Y.; Ye, X.; Zhang, H.M.; Hanson, P.; Zhao, G.; Tong, L.; Xie, R.; Yang, D. Cleavage of osmosensitive transcriptional factor NFAT5 by Coxsackieviral protease 2A promotes viral replication. PLoS Pathog. 2017, 13, e1006744. [Google Scholar] [CrossRef]
- Xie, L.; Lu, B.; Zheng, Z.; Miao, Y.; Liu, Y.; Zhang, Y.; Zheng, C.; Ke, X.; Hu, Q.; Wang, H. The 3C protease of enterovirus A71 counteracts the activity of host zinc-finger antiviral protein (ZAP). J. Gen. Virol. 2018, 99, 73–85. [Google Scholar] [CrossRef]
- Marcotte, L.L.; Wass, A.B.; Gohara, D.W.; Pathak, H.B.; Arnold, J.J.; Filman, D.J.; Cameron, C.E.; Hogle, J.M. Crystal Structure of Poliovirus 3CD Protein: Virally Encoded Protease and Precursor to the RNA-Dependent RNA Polymerase. J. Virol. 2007, 81, 3583–3596. [Google Scholar] [CrossRef]
- Palmenberg, A.C. Picornaviral processing: Some new ideas. J. Cell. Biochem. 1987, 33, 191–198. [Google Scholar] [CrossRef]
- Kim, B.-K.; Cho, J.-H.; Jeong, P.; Lee, Y.; Lim, J.J.; Park, K.R.; Eom, S.H.; Kim, Y.-C. Benserazide, the first allosteric inhibitor of Coxsackievirus B3 3C protease. FEBS Lett. 2015, 589, 1795–1801. [Google Scholar] [CrossRef]
- Lim, B.-K.; Yun, S.-H.; Ju, E.-S.; Kim, B.-K.; Lee, Y.-J.; Yoo, D.-K.; Kim, Y.-C.; Jeon, E.-S. Soluble coxsackievirus B3 3C protease inhibitor prevents cardiomyopathy in an experimental chronic myocarditis murine model. Virus Res. 2015, 199, 1–8. [Google Scholar] [CrossRef]
- Kim, B.-K.; Ko, H.; Jeon, E.-S.; Ju, E.-S.; Jeong, L.S.; Kim, Y.-C. 2,3,4-Trihydroxybenzyl-hydrazide analogues as novel potent coxsackievirus B3 3C protease inhibitors. Eur. J. Med. Chem. 2016, 120, 202–216. [Google Scholar] [CrossRef]
- Gerner, E.W.; Meyskens, F.L. Polyamines and cancer: Old molecules, new understanding. Nat. Rev. Cancer 2004, 4, 781–792. [Google Scholar] [CrossRef]
- Frugier, M.; Florentz, C.; Hosseini, M.W.; Lehn, J.M.; Giegé, R. Synthetic polyamines stimulate in vitro transcription by T7 RNA polymerase. Nucleic Acids Res. 1994, 22, 2784–2790. [Google Scholar] [CrossRef]
- Mandal, S.; Mandal, A.; Johansson, H.E.; Orjalo, A.V.; Park, M.H. Depletion of cellular polyamines, spermidine and spermine, causes a total arrest in translation and growth in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 2169–2174. [Google Scholar] [CrossRef]
- Roberts, S.; Ullman, B. Parasite Polyamines as Pharmaceutical Targets. Curr. Pharm. Des. 2017, 23, 3325–3341. [Google Scholar] [CrossRef]
- Alexiou, G.A.; Lianos, G.D.; Ragos, V.; Galani, V.; Kyritsis, A.P. Difluoromethylornithine in cancer: New advances. Future Oncol. Lond. Engl. 2017, 13, 809–819. [Google Scholar] [CrossRef]
- Gerner, E.W.; Bruckheimer, E.; Cohen, A. Cancer pharmacoprevention: Targeting polyamine metabolism to manage risk factors for colon cancer. J. Biol. Chem. 2018, 293, 18770–18778. [Google Scholar] [CrossRef]
- Fabian, C.J.; Kimler, B.F.; Brady, D.A.; Mayo, M.S.; Chang, C.H.J.; Ferraro, J.A.; Zalles, C.M.; Stanton, A.L.; Masood, S.; Grizzle, W.E.; et al. A phase II breast cancer chemoprevention trial of oral alpha-difluoromethylornithine: Breast tissue, imaging, and serum and urine biomarkers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 3105–3117. [Google Scholar]
- Simoneau, A.R.; Gerner, E.W.; Nagle, R.; Ziogas, A.; Fujikawa-Brooks, S.; Yerushalmi, H.; Ahlering, T.E.; Lieberman, R.; McLaren, C.E.; Anton-Culver, H.; et al. The effect of difluoromethylornithine on decreasing prostate size and polyamines in men: Results of a year-long phase IIb randomized placebo-controlled chemoprevention trial. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2008, 17, 292–299. [Google Scholar] [CrossRef]
- Mounce, B.C.; Olsen, M.E.; Vignuzzi, M.; Connor, J.H. Polyamines and Their Role in Virus Infection. Microbiol. Mol. Biol. Rev. MMBR 2017, 81, e00029-17. [Google Scholar] [CrossRef]
- Mounce, B.C.; Cesaro, T.; Moratorio, G.; Hooikaas, P.J.; Yakovleva, A.; Werneke, S.W.; Smith, E.C.; Poirier, E.Z.; Simon-Loriere, E.; Prot, M.; et al. Inhibition of Polyamine Biosynthesis Is a Broad-Spectrum Strategy against RNA Viruses. J. Virol. 2016, 90, 9683–9692. [Google Scholar] [CrossRef]
- Mounce, B.C.; Poirier, E.Z.; Passoni, G.; Simon-Loriere, E.; Cesaro, T.; Prot, M.; Stapleford, K.A.; Moratorio, G.; Sakuntabhai, A.; Levraud, J.-P.; et al. Interferon-Induced Spermidine-Spermine Acetyltransferase and Polyamine Depletion Restrict Zika and Chikungunya Viruses. Cell Host Microbe 2016, 20, 167–177. [Google Scholar] [CrossRef]
- Cai, Q.; Yameen, M.; Liu, W.; Gao, Z.; Li, Y.; Peng, X.; Cai, Y.; Wu, C.; Zheng, Q.; Li, J.; et al. Conformational Plasticity of the 2A Proteinase from Enterovirus 71. J. Virol. 2013, 87, 7348–7356. [Google Scholar] [CrossRef]
- Kandolf, R.; Hofschneider, P.H. Molecular cloning of the genome of a cardiotropic Coxsackie B3 virus: Full-length reverse-transcribed recombinant cDNA generates infectious virus in mammalian cells. Proc. Natl. Acad. Sci. USA 1985, 82, 4818–4822. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Madhubala, R. Thin-layer chromatographic method for assaying polyamines. Methods Mol. Biol. 1998, 79, 131–136. [Google Scholar]
- Mounce, B.C.; Cesaro, T.; Vlajnić, L.; Vidiņa, A.; Vallet, T.; Weger-Lucarelli, J.; Passoni, G.; Stapleford, K.A.; Levraud, J.-P.; Vignuzzi, M. Chikungunya Virus Overcomes Polyamine Depletion by Mutation of nsP1 and the Opal Stop Codon To Confer Enhanced Replication and Fitness. J. Virol. 2017, 91, e00344-17. [Google Scholar] [CrossRef]
- Farhadian, S.; Shareghi, B.; Saboury, A.A. Exploring the thermal stability and activity of α-chymotrypsin in the presence of spermine. J. Biomol. Struct. Dyn. 2017, 35, 435–448. [Google Scholar] [CrossRef]
- Rezaei-Ghaleh, N.; Ebrahim-Habibi, A.; Moosavi-Movahedi, A.A.; Nemat-Gorgani, M. Effect of polyamines on the structure, thermal stability and 2,2,2-trifluoroethanol-induced aggregation of alpha-chymotrypsin. Int. J. Biol. Macromol. 2007, 41, 597–604. [Google Scholar] [CrossRef]
- Kilianski, A.; Mielech, A.M.; Deng, X.; Baker, S.C. Assessing activity and inhibition of Middle East respiratory syndrome coronavirus papain-like and 3C-like proteases using luciferase-based biosensors. J. Virol. 2013, 87, 11955–11962. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhong, X.; Lin, L.; Wu, S.; Wang, T.; Chen, Y.; Zhai, X.; Wang, Y.; Wu, H.; Tong, L.; et al. A 3C(pro)-dependent bioluminescence imaging assay for in vivo evaluation of anti-enterovirus 71 agents. Antiviral Res. 2014, 101, 82–92. [Google Scholar] [CrossRef]
- Wang, L.; Fu, Q.; Dong, Y.; Zhou, Y.; Jia, S.; Du, J.; Zhao, F.; Wang, Y.; Wang, X.; Peng, J.; et al. Bioluminescence imaging of Hepatitis C virus NS3/4A serine protease activity in cells and living animals. Antiviral Res. 2010, 87, 50–56. [Google Scholar] [CrossRef]
- Blom, N.; Hansen, J.; Brunak, S.; Blaas, D. Cleavage site analysis in picornaviral polyproteins: Discovering cellular targets by neural networks. Protein Sci. 1996, 5, 2203–2216. [Google Scholar] [CrossRef]
- Lee, C.-C.; Kuo, C.-J.; Ko, T.-P.; Hsu, M.-F.; Tsui, Y.-C.; Chang, S.-C.; Yang, S.; Chen, S.-J.; Chen, H.-C.; Hsu, M.-C.; et al. Structural Basis of Inhibition Specificities of 3C and 3C-like Proteases by Zinc-coordinating and Peptidomimetic Compounds. J. Biol. Chem. 2009, 284, 7646–7655. [Google Scholar] [CrossRef] [PubMed]
- Kaur-Sawhney, R.; Shih, L.; Cegielska, T.; Galston, A.W. Inhibition of protease activity by polyamines. FEBS Lett. 1982, 145, 345–349. [Google Scholar] [CrossRef]
- Balestreri, E.; Cioni, P.; Romagnoli, A.; Bernini, S.; Fissi, A.; Felicioli, R. Mechanism of polyamine inhibition of a leaf protease. Arch. Biochem. Biophys. 1987, 255, 460–463. [Google Scholar] [CrossRef]
- Shih, L.-M.; Kaur-Sawhney, R.; Fuhrer, J.; Samanta, S.; Galston, A.W. Effects of Exogenous 1,3-Diaminopropane and Spermidine on Senescence of Oat Leaves: I. Inhibition of Protease Activity, Ethylene Production, and Chlorophyll Loss as Related to Polyamine Content. PLANT Physiol. 1982, 70, 1592–1596. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protease/Target | Function | Sense | Sequence |
|---|---|---|---|
| 2A | Clone | F | 5’-GCTGCGGCCGCAGGCGCATTTGGACAACAATCAGG-3’ |
| Clone | R | 5’-GCTTCTAGATTACTGTTCCATTGCATCATCCTTCCAG-3’ | |
| Sequence | F | 5’-CATGTCAAAGCGTGGATACCTAGAC-3’ | |
| Sequence | R | 5’-GCACATGGGATTGGTATCTCCTGGG-3’ | |
| qPCR | F | 5’-CATGTCAAAGCGTGGATACCTAGAC-3’ | |
| qPCR | R | 5’-GCACATGGGATTGGTATCTCCTGGG-3’ | |
| 3C | Clone | F | 5’-GTTGCGGCCGCTGGCCCTGCCTTTGAGTTCGCCG-3’ |
| Clone | R | 5’-GCGTCTAGATTATTGCTCATCATTGAAGTAGTGTTTG-3’ | |
| Sequence | F | 5’-TATACAGGAGTGCCCAACCAGAAGC-3’ | |
| Sequence | R | 5’-GAATGTACATGTTGGGAAACTTGCT-3’ | |
| qPCR | F | 5’-AGGGCGAGATCAATCACATTAG-3’ | |
| qPCR | R | 5’-CTCTGCTGTTGCCTCACTATC-3’ | |
| 2A ProTarget | Clone | F | 5’-GATCCATGACCAACACCGGCGCGTTTGGCTAA-3’ |
| Clone | R | 5’-AGCTTTAGCCAAACGCGCCGGTGTTGGTCATG-3’ | |
| 3C ProTarget | Clone | F | 5’-GATCCGCGATGGAACAGGGCTAA-3’ |
| Clone | R | 5’-AGCTTTAGCCCTGTTCCATCGCG-3’ | |
| 2A/3C ProTarget | Sequence | F | 5’-GTAGTACAGACTGGAAAATATC-3’ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dial, C.N.; Tate, P.M.; Kicmal, T.M.; Mounce, B.C. Coxsackievirus B3 Responds to Polyamine Depletion via Enhancement of 2A and 3C Protease Activity. Viruses 2019, 11, 403. https://doi.org/10.3390/v11050403
Dial CN, Tate PM, Kicmal TM, Mounce BC. Coxsackievirus B3 Responds to Polyamine Depletion via Enhancement of 2A and 3C Protease Activity. Viruses. 2019; 11(5):403. https://doi.org/10.3390/v11050403
Chicago/Turabian StyleDial, Courtney N., Patrick M. Tate, Thomas M. Kicmal, and Bryan C. Mounce. 2019. "Coxsackievirus B3 Responds to Polyamine Depletion via Enhancement of 2A and 3C Protease Activity" Viruses 11, no. 5: 403. https://doi.org/10.3390/v11050403
APA StyleDial, C. N., Tate, P. M., Kicmal, T. M., & Mounce, B. C. (2019). Coxsackievirus B3 Responds to Polyamine Depletion via Enhancement of 2A and 3C Protease Activity. Viruses, 11(5), 403. https://doi.org/10.3390/v11050403