Prevalence and Diversity Analysis of Candidate Prophages to Provide An Understanding on Their Roles in Bacillus Thuringiensis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of the Bt Genome Dataset and Prediction of Prophage Regions
2.2. Bioinformatic Analysis of Prophage Sequences
2.3. Induction and Characteristic of Prophages from Genome-Sequenced Bt Strains
2.4. In-Gel Lytic Activity Assay
3. Results
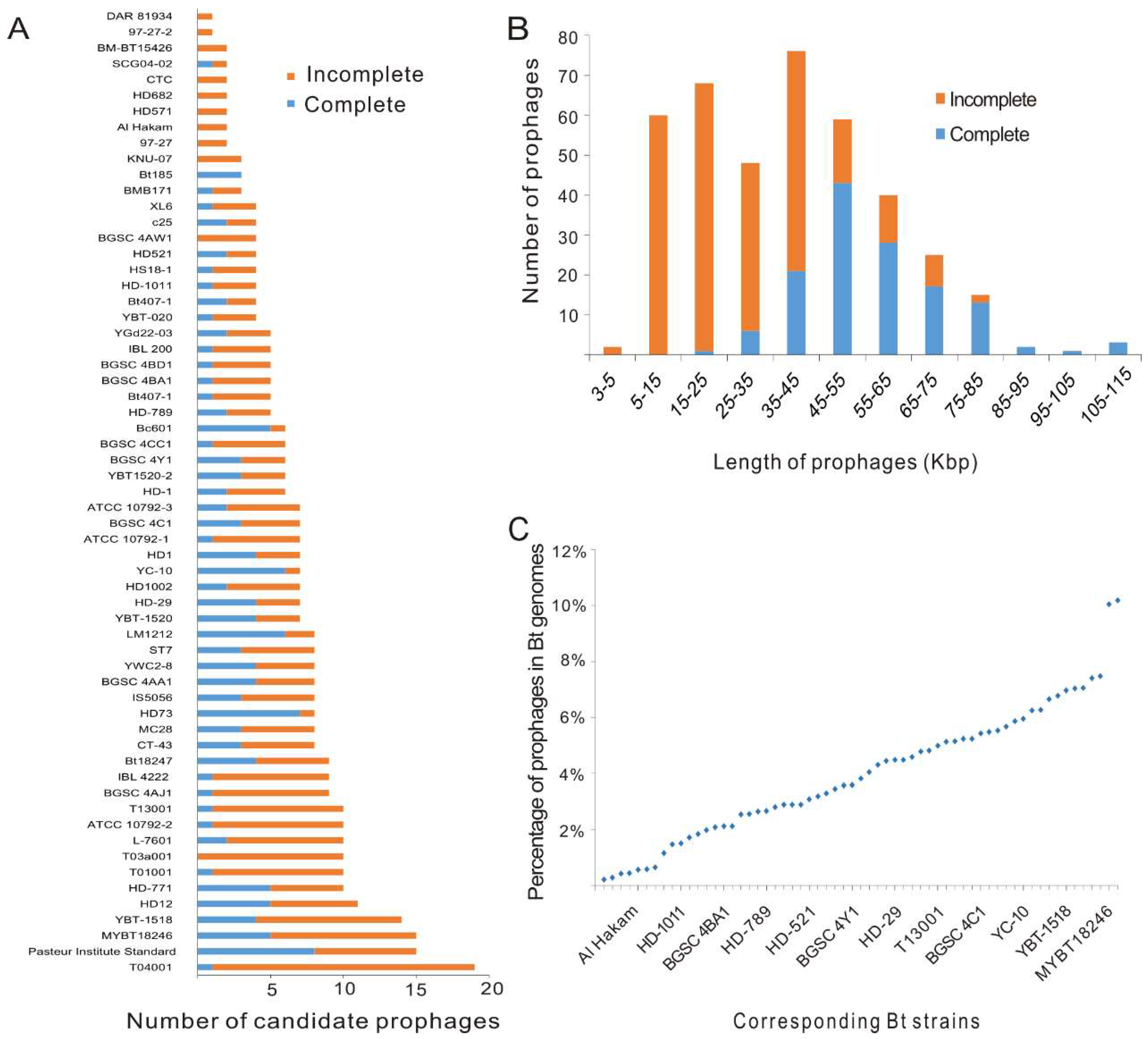
3.1. High Prevalence of Prophage Sequences in Bt Genomes
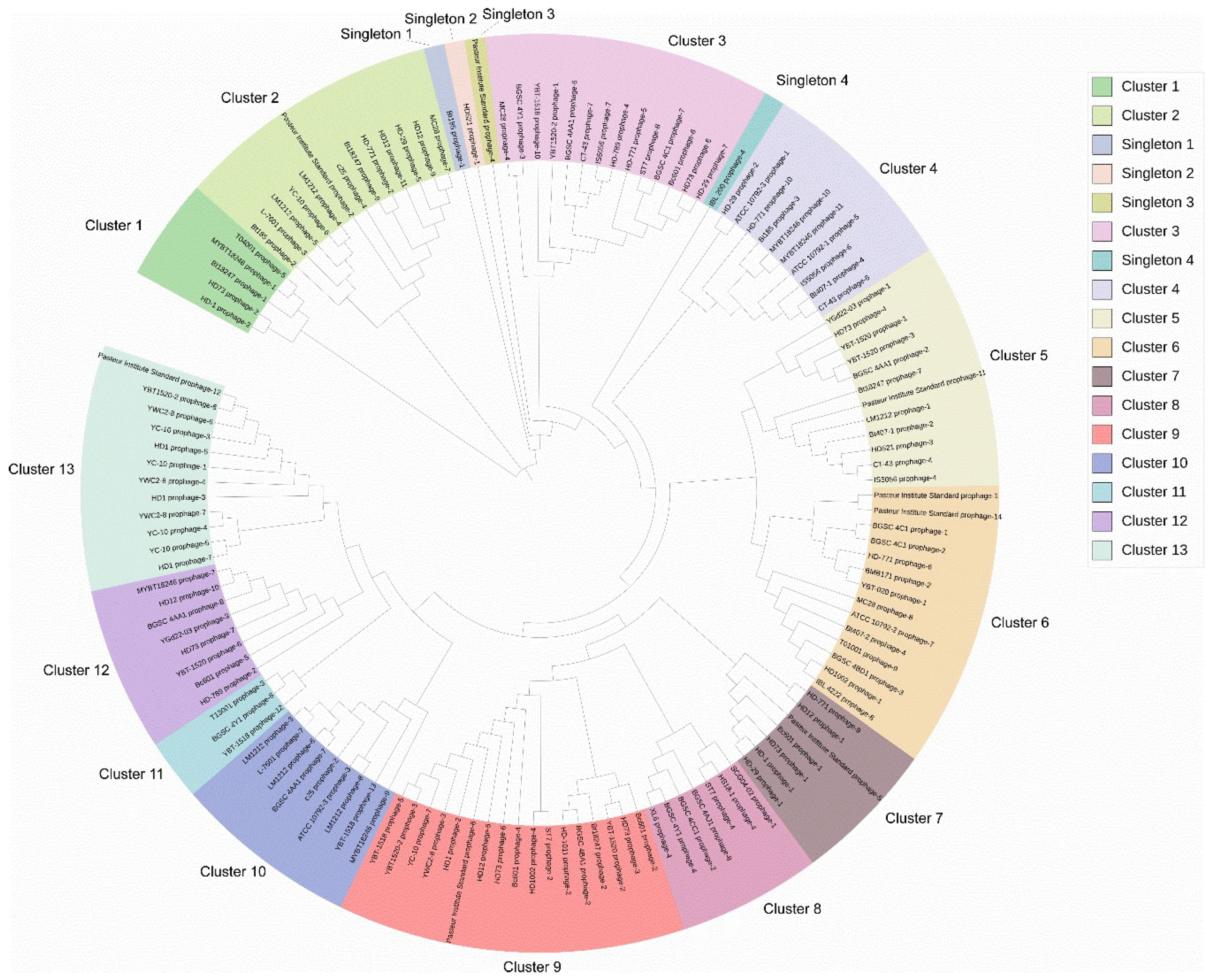
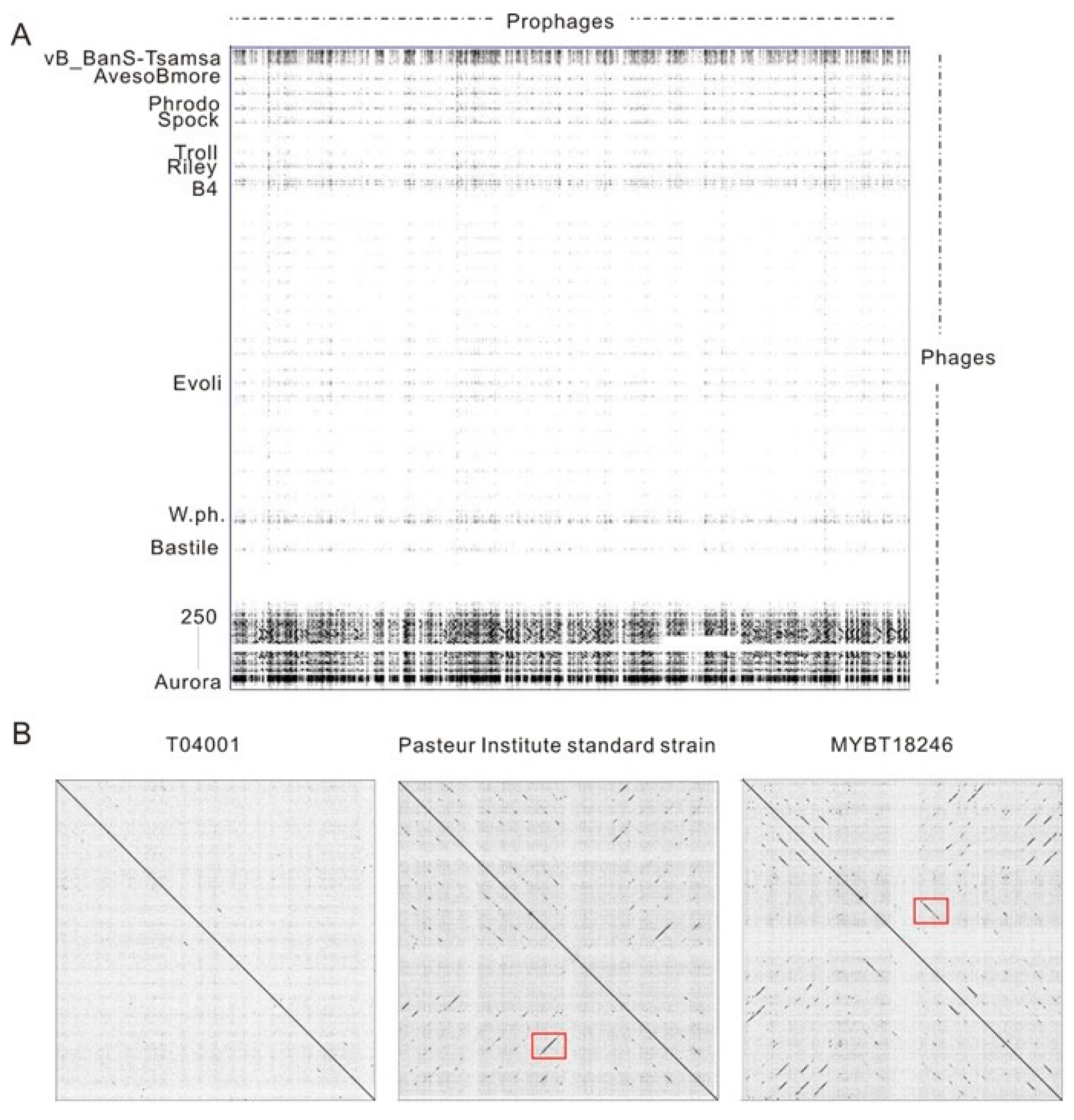
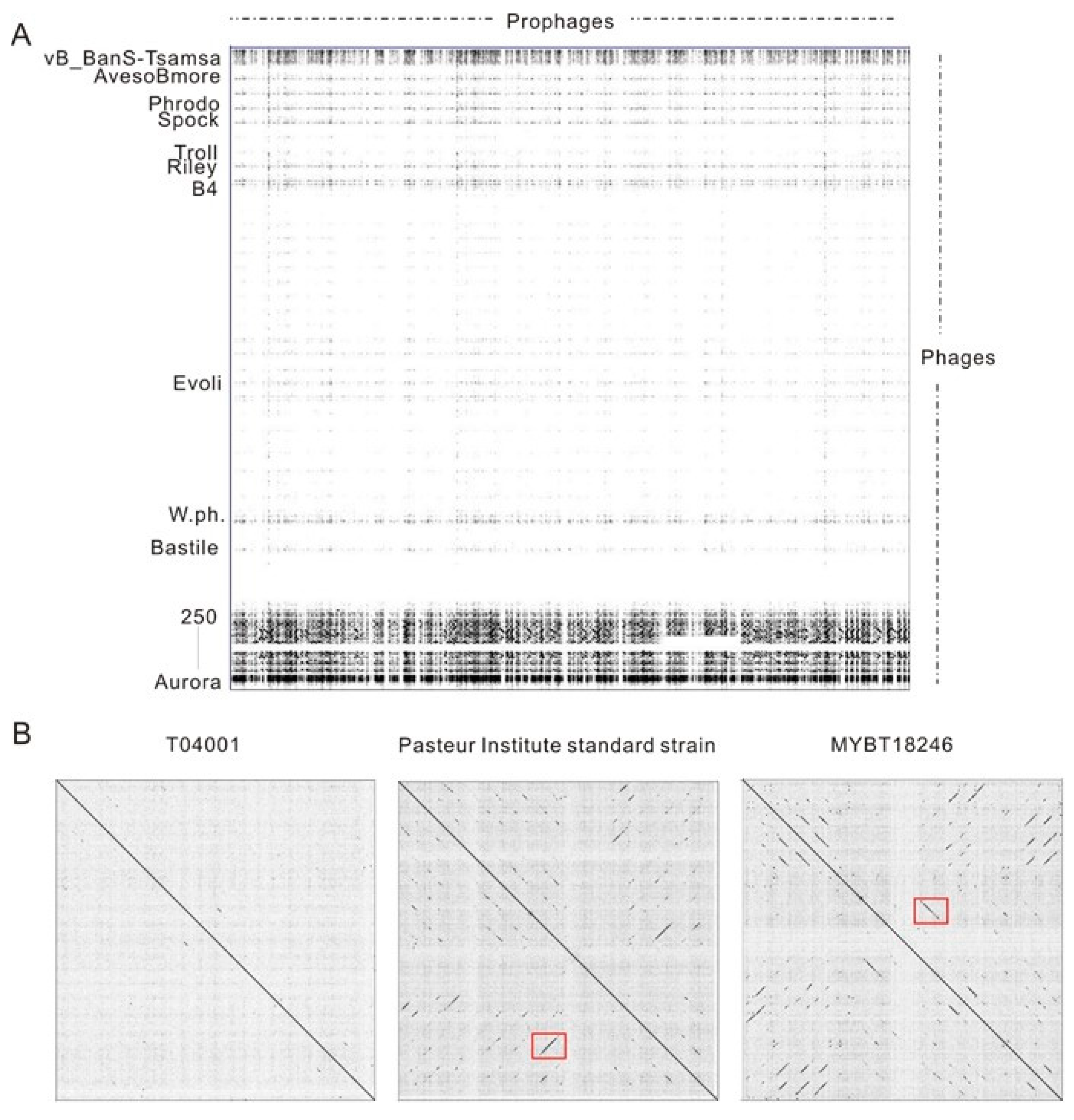
3.2. Diversity and Phylogenetic Relationship Analysis of Putative Complete Prophages from Bt Genomes
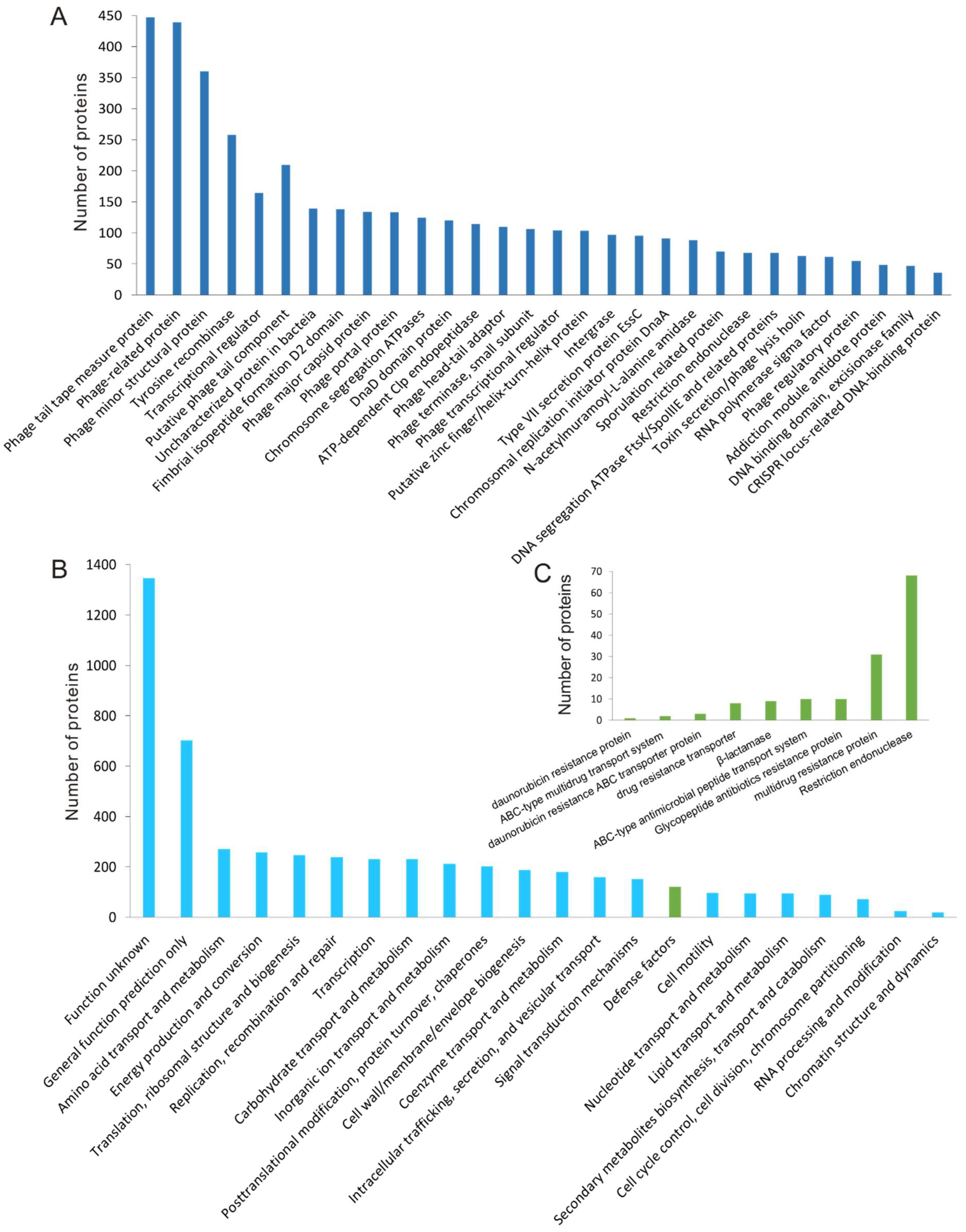
3.3. Bioinformatic Analysis of Proteins Encoded by Bt Prophages
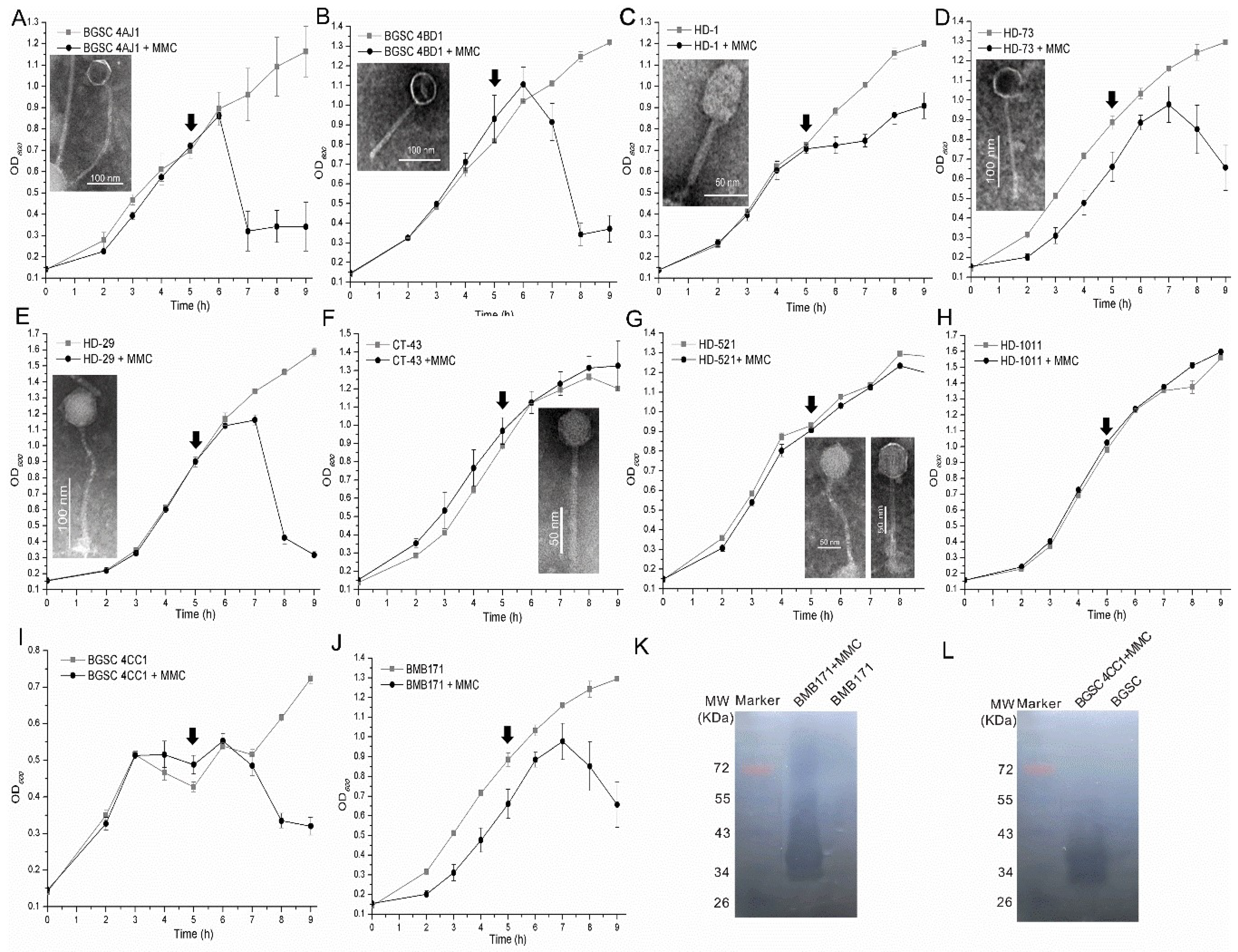
3.4. High Inducibility of Bt Prophages
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bravo, A.; Likitvivatanavong, S.; Gill, S.S.; Soberon, M. Bacillus thuringiensis: A story of a successful bioinsecticide. Insect Biochem. Mol. Biol. 2011, 41, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Jouzani, G.S.; Valijanian, E.; Sharafi, R. Bacillus thuringiensis: A successful insecticide with new environmental features and tidings. Appl. Microbiol. Biotechnol. 2017, 101, 2691–2711. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Song, S.Y.; Sun, F.; Jia, Y.H.; Zeng, W.H.; Pang, Y. Isolation, characterization and genome sequencing of phage MZTP02 from Bacillus thuringiensis MZ1. Arch. Virol. 2008, 153, 1855–1865. [Google Scholar] [CrossRef]
- Gillis, A.; Mahillon, J. Influence of Lysogeny of Tectiviruses GIL01 and GIL16 on Bacillus thuringiensis Growth, Biofilm Formation, and Swarming Motility. Appl. Environ. Microbiol. 2014, 80, 7620–7630. [Google Scholar] [CrossRef] [PubMed]
- Gillis, A.; Mahillon, J. Phages Preying on Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis: Past, Present and Future. Viruses 2014, 6, 2623–2672. [Google Scholar] [CrossRef] [PubMed]
- Sauder, A.B.; Quinn, M.R.; Brouillette, A.; Caruso, S.; Cresawn, S.; Erill, I.; Lewis, L.; Loesser-Casey, K.; Pate, M.; Scott, C.; et al. Genomic characterization and comparison of seven Myoviridae bacteriophage infecting Bacillus thuringiensis. Virology 2016, 489, 243–251. [Google Scholar] [CrossRef]
- Wu, D.D.; Yuan, Y.H.; Liu, P.M.; Wu, Y.; Gao, M.Y. Cellular responses in Bacillus thuringiensis CS33 during bacteriophage BtCS33 infection. J. Proteomics 2014, 101, 192–204. [Google Scholar] [CrossRef]
- Casjens, S. Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 2003, 49, 277–300. [Google Scholar] [CrossRef]
- Ikeda, H.; Tomizawa, J. Prophage P1, and extrachromosomal replication unit. Cold Spring Harb. Symp. Quant. Biol. 1968, 33, 791–798. [Google Scholar] [CrossRef]
- Ravin, V.K.; Shulga, M.G. Evidence for extrachromosomal location of prophage N15. Virology 1970, 40, 800–807. [Google Scholar] [CrossRef]
- Girons, I.S.; Bourhy, P.; Ottone, C.; Picardeau, M.; Yelton, D.; Hendrix, R.W.; Glaser, P.; Charon, N. The LE1 bacteriophage replicates as a plasmid within Leptospira biflexa: Construction of an L. biflexa-Escherichia coli shuttle vector. J. Bacteriol. 2000, 182, 5700–5705. [Google Scholar] [CrossRef] [PubMed]
- Kalckar, H.M.; Sundararajanta. Regulatory mechanisms in the synthesis of enzymes of galactose metabolism. II. Genetic defects in galactokinase activity and their relations to its function. Cold Spring Harb. Symp. Quant. Biol. 1961, 26, 227–231. [Google Scholar] [CrossRef]
- Banuett, F.; Herskowitz, I. Identification of polypeptides encoded by an Escherichia coli locus (hflA) that governs the lysis-lysogeny decision of bacteriophage lambda. J. Bacteriol. 1987, 169, 4076–4085. [Google Scholar] [CrossRef] [PubMed]
- Engelberg-Kulka, H.; Kumar, S. Yet another way that phage manipulates its Escherichia coli host: rexB is involved in the lysogenic-lytic switch. Mol. Microbiol. 2015, 96, 689–693. [Google Scholar] [CrossRef]
- Hammerl, J.A.; Jackel, C.; Funk, E.; Pinnau, S.; Mache, C.; Hertwig, S. The diverse genetic switch of enterobacterial and marine telomere phages. Bacteriophage 2016, 6, e1148805. [Google Scholar] [CrossRef] [PubMed]
- Broussard, G.W.; Oldfield, L.M.; Villanueva, V.M.; Lunt, B.L.; Shine, E.E.; Hatfull, G.F. Integration-Dependent Bacteriophage Immunity Provides Insights into the Evolution of Genetic Switches. Mol. Cell 2013, 49, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W.; Azizbekyan, R.R.; Konjin, H.P.E.; Lecadet, M.M.; Seldin, L.; Yu, M.X. New Bacillus Bacteriophage Species. Arch. Virol. 1994, 135, 333–344. [Google Scholar] [CrossRef]
- Colasito, D.J.; Rogoff, M.H. Characterization of Temperate Bacteriophages of Bacillus Thuringiensis. J. Gen. Virol. 1969, 5, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Azizbekian, R.R.; Smirnova, T.A.; Minenkova, I.B.; Rebentish, B.A. Lysogeny in Bacillus thuringiensis. Mikrobiologiia 1980, 49, 961–968. [Google Scholar] [PubMed]
- Kochkina, Z.M. Cleavage of DNA from 2 phages of Bacillus thuringiensis by EcoRI and HindIII endonucleases. Mikrobiologiia 1986, 55, 1045–1047. [Google Scholar] [PubMed]
- Smirnova, T.A.; Minenkova, I.B.; Rebentish, B.A.; Azizbekian, R.R. Characteristics of Bacillus thuringiensis phages with circular permutation in the DNA molecule. Mol. Gen. Mikrobiol. Virusol. 1985, 6, 21–23. [Google Scholar]
- Dong, Z.; Peng, D.; Wang, Y.; Zhu, L.; Ruan, L.; Sun, M. Complete Genome Sequence of Bacillus thuringiensis Bacteriophage BMBtp2. Genome Announc. 2013, 1. [Google Scholar] [CrossRef]
- Roh, J.Y.; Bin Park, J.; Liu, Q.; Kim, S.E.; Tao, X.; Choi, T.W.; Choi, J.Y.; Kim, W.J.; Jin, B.R.; Je, Y.H. Existence of lysogenic bacteriophages in Bacillus thuringiensis type strains. J. Invertebr. Pathol. 2013, 113, 228–231. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Jacobs-Sera, D.; Bustamante, C.A.; Garlena, R.A.; Mavrich, T.N.; Pope, W.H.; Reyes, J.C.; Russell, D.A.; Adair, T.; Alvey, R.; et al. Prophage-mediated defence against viral attack and viral counter-defence. Nat. Microbiol. 2017, 2, 16251. [Google Scholar] [CrossRef]
- Brueggemann, A.B.; Harrold, C.L.; Rezaei Javan, R.; van Tonder, A.J.; McDonnell, A.J.; Edwards, B.A. Pneumococcal prophages are diverse, but not without structure or history. Sci. Rep. 2017, 7, 42976. [Google Scholar] [CrossRef] [PubMed]
- Crispim, J.S.; Dias, R.S.; Vidigal, P.M.P.; de Sousa, M.P.; da Silva, C.C.; Santana, M.F.; de Paula, S.O. Screening and characterization of prophages in Desulfovibrio genomes. Sci. Rep. 2018, 8, 9273. [Google Scholar] [CrossRef]
- Asadulghani, M.; Ogura, Y.; Ooka, T.; Itoh, T.; Sawaguchi, A.; Iguchi, A.; Nakayama, K.; Hayashi, T. The defective prophage pool of Escherichia coli O157: Prophage-prophage interactions potentiate horizontal transfer of virulence determinants. PLoS Pathog. 2009, 5, e1000408. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.J.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Krumsiek, J.; Arnold, R.; Rattei, T. Gepard: A rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 2007, 23, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 2001, 29, 2607–2618. [Google Scholar] [CrossRef]
- Wu, S.T.; Zhu, Z.W.; Fu, L.M.; Niu, B.F.; Li, W.Z. WebMGA: A customizable web server for fast metagenomic sequence analysis. BMC Genomics 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.Y.; Li, R.S.; Dai, S.Y.; Wu, Y.; Yi, D. Diversity of Bacillus thuringiensis strains from soil in China and their pesticidal activities. Biol. Control 2008, 44, 380–388. [Google Scholar] [CrossRef]
- Yamamoto, K.R.; Alberts, B.M.; Benzinger, R.; Lawhorne, L.; Treiber, G. Rapid bacteriophage sedimentation in the presence of polyethylene glycol and its application to large-scale virus purification. Virology 1970, 40, 734–744. [Google Scholar] [CrossRef]
- Yuan, Y.H.; Gao, M.Y.; Wu, D.D.; Liu, P.M.; Wu, Y. Genome Characteristics of a Novel Phage from Bacillus thuringiensis Showing High Similarity with Phage from Bacillus cereus. Plos ONE 2012, 7, e37557. [Google Scholar] [CrossRef]
- Yuan, Y.H.; Gao, M.Y.; Peng, Q.; Wu, D.D.; Liu, P.M.; Wu, Y. Genomic analysis of a phage and prophage from a Bacillus thuringiensis strain. J. Gen. Virol. 2014, 95, 751–761. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genomics 2008, 9. [Google Scholar] [CrossRef]
- Besemer, J.; Borodovsky, M. GeneMark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005, 33, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- Soding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.M.; Chetvernin, V.; Tatusova, T. PAirwise Sequence Comparison (PASC) and Its Application in the Classification of Filoviruses. Viruses 2012, 4, 1318–1327. [Google Scholar] [CrossRef]
- Yuan, Y.; Gao, M. Proteomic Analysis of a Novel Bacillus Jumbo Phage Revealing Glycoside Hydrolase As Structural Component. Front. Microbiol. 2016, 7, 745. [Google Scholar] [CrossRef] [PubMed]
- Tock, M.R.; Dryden, D.T.F. The biology of restriction and anti-restriction. Current opinion in microbiology 2005, 8, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Kim, Y.; Ma, Q.; Hong, S.H.; Pokusaeva, K.; Sturino, J.M.; Wood, T.K. Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.N.; Zhang, J.Y.; Xu, J.L.; Du, P.C.; Pang, B.; Li, J.; Kan, B. The Resistance of Vibrio cholerae O1 El Tor Strains to the Typing Phage 919TP, a Member of K139 Phage Family. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Qian, J.; Westra, E.R.; Buckling, A.; Guttman, D.S.; Davidson, A.R.; Maxwell, K.L. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016, 10, 2854–2866. [Google Scholar] [CrossRef] [Green Version]
- Low, L.Y.; Yang, C.; Perego, M.; Osterman, A.; Liddington, R.C. Structure and lytic activity of a Bacillus anthracis prophage endolysin. J. Biol. Chem. 2005, 280, 35433–35439. [Google Scholar] [CrossRef]
- Regamey, A.; Karamata, D. The N-acetylmuramoyl-L-alanine amidase encoded by the Bacillus subtilis 168 prophage SP beta. Microbiology 1998, 144 ( Pt 4), 885–893. [Google Scholar] [CrossRef]
- Bae, T.; Baba, T.; Hiramatsu, K.; Schneewind, O. Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol. Microbiol. 2006, 62, 1035–1047. [Google Scholar] [CrossRef]
- Matos, R.C.; Lapaque, N.; Rigottier-Gois, L.; Debarbieux, L.; Meylheuc, T.; Gonzalez-Zorn, B.; Repoila, F.; Lopes Mde, F.; Serror, P. Enterococcus faecalis prophage dynamics and contributions to pathogenic traits. PLoS Genet. 2013, 9, e1003539. [Google Scholar] [CrossRef]
- Lee, Y.D.; Park, J.H. Phage Conversion for beta-Lactam Antibiotic Resistance of Staphylococcus aureus from Foods. J. Microbiol. Biotechnol. 2016, 26, 263–269. [Google Scholar] [CrossRef]
- Doron, S.; Melamed, S.; Ofir, G.; Leavitt, A.; Lopatina, A.; Keren, M.; Amitai, G.; Sorek, R. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 2018, 359. [Google Scholar] [CrossRef]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Cumby, N.; Edwards, A.M.; Davidson, A.R.; Maxwell, K.L. The bacteriophage HK97 gp15 moron element encodes a novel superinfection exclusion protein. J. Bacteriol. 2012, 194, 5012–5019. [Google Scholar] [CrossRef]
- Uc-Mass, A.; Loeza, E.J.; de la Garza, M.; Guarneros, G.; Hernandez-Sanchez, J.; Kameyama, L. An orthologue of the cor gene is involved in the exclusion of temperate lambdoid phages. Evidence that Cor inactivates FhuA receptor functions. Virology 2004, 329, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Gohler, A.; Heller, K.J.; Neve, H. The ltp gene of temperate Streptococcus thermophilus phage TP-J34 confers superinfection exclusion to Streptococcus thermophilus and Lactococcus lactis. Virology 2006, 350, 146–157. [Google Scholar] [CrossRef]
- Fortier, L.C.; Sekulovic, O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 2013, 4, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Schnepf, E.; Crickmore, N.; Van Rie, J.; Lereclus, D.; Baum, J.; Feitelson, J.; Zeigler, D.R.; Dean, D.H. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol. Mol. Biol. Rev. 1998, 62, 775–806. [Google Scholar] [PubMed]
- Isom, C.E.; Menon, S.K.; Thomas, L.M.; West, A.H.; Richter-Addo, G.B.; Karr, E.A. Crystal structure and DNA binding activity of a PadR family transcription regulator from hypervirulent Clostridium difficile R20291. BMC Microbiol. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Quiles-Puchalt, N.; Tormo-Mas, M.A.; Campoy, S.; Toledo-Arana, A.; Monedero, V.; Lasa, I.; Novick, R.P.; Christie, G.E.; Penades, J.R. A super-family of transcriptional activators regulates bacteriophage packaging and lysis in Gram-positive bacteria. Nucleic Acids Res. 2013, 41, 7260–7275. [Google Scholar] [CrossRef] [Green Version]
- Goto, N.; Mochizuki, A.; Inagaki, Y.; Horiuchi, S.; Tanaka, T.; Nakaya, R. Identification of the DNA-Sequence Required for Transposition Immunity of the Gamma-Delta-Sequence. J. Bacteriol. 1987, 169, 4388–4390. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Bhagwat, A.S.; Heffron, F. Identification of a Transposon-Tn3 Sequence Required for Transposition Immunity. Proc. Natl. Acad. Sci. USA 1983, 80, 6765–6769. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Dramicanin, M.; Mizuuchi, M.; Adam, J.; Wang, Y.; Han, Y.W.; Yang, W.; Steven, A.C.; Mizuuchi, K.; Ramon-Maiques, S. MuB is an AAA plus ATPase that forms helical filaments to control target selection for DNA transposition. Proc. Natl. Acad. Sci. USA 2013, 110, E2441–E2450. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Y.; Wu, Y.; Yuan, Y.; Gao, M. Prevalence and Diversity Analysis of Candidate Prophages to Provide An Understanding on Their Roles in Bacillus Thuringiensis. Viruses 2019, 11, 388. https://doi.org/10.3390/v11040388
Fu Y, Wu Y, Yuan Y, Gao M. Prevalence and Diversity Analysis of Candidate Prophages to Provide An Understanding on Their Roles in Bacillus Thuringiensis. Viruses. 2019; 11(4):388. https://doi.org/10.3390/v11040388
Chicago/Turabian StyleFu, Yajuan, Yan Wu, Yihui Yuan, and Meiying Gao. 2019. "Prevalence and Diversity Analysis of Candidate Prophages to Provide An Understanding on Their Roles in Bacillus Thuringiensis" Viruses 11, no. 4: 388. https://doi.org/10.3390/v11040388
APA StyleFu, Y., Wu, Y., Yuan, Y., & Gao, M. (2019). Prevalence and Diversity Analysis of Candidate Prophages to Provide An Understanding on Their Roles in Bacillus Thuringiensis. Viruses, 11(4), 388. https://doi.org/10.3390/v11040388