Identification of a Novel Gammaherpesvirus in Canada lynx (Lynx canadensis)

 , ,
, ,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Preparation
2.2. Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) PCR
2.3. Amplification of GHV Glycoprotein B (gB) Sequence Using Degenerate Primers
2.4. Amplification of GHV DNA Polymerase (DNApol) Sequence Using Degenerate Primers
2.5. Long-Distance PCR and Sequencing
2.6. LcaGHV1 Detection PCR Assay
2.7. Statistical Analysis
2.8. Phylogenetic Analysis
2.9. Nucleotide Sequence Accession Numbers
3. Results
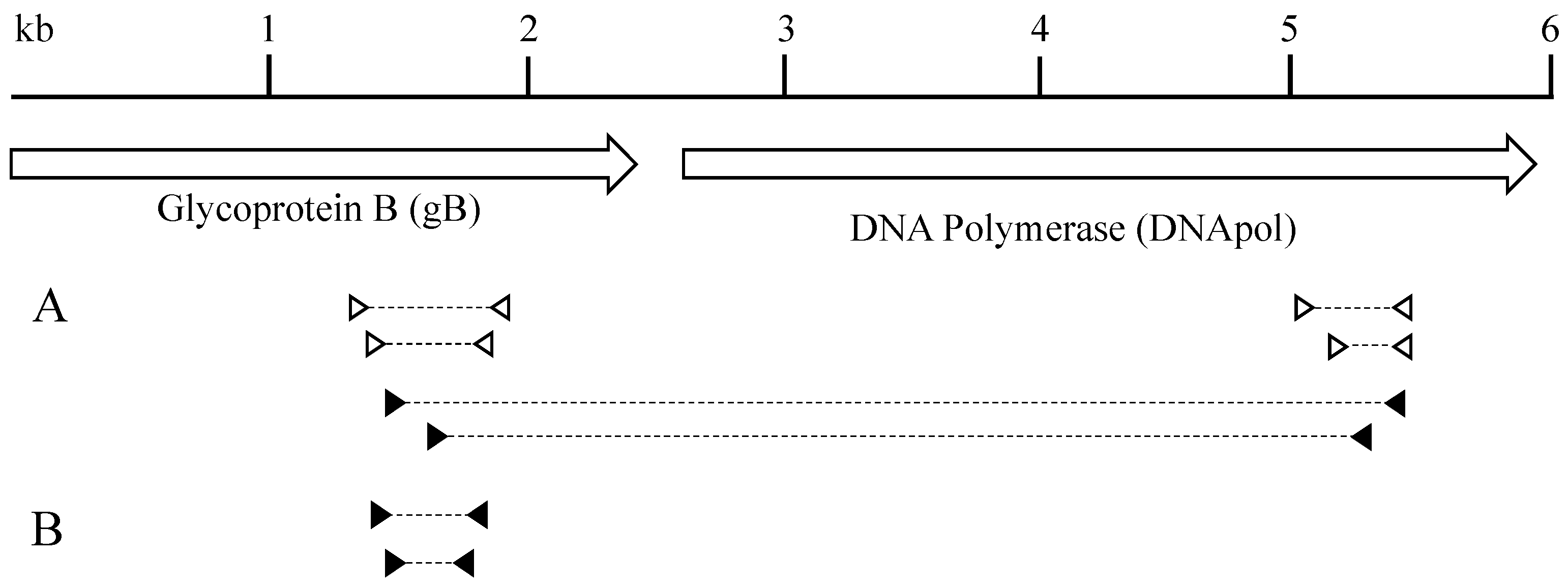
3.1. Detection of a Novel Gammaherpesvirus Sequence in Lynx Tissues
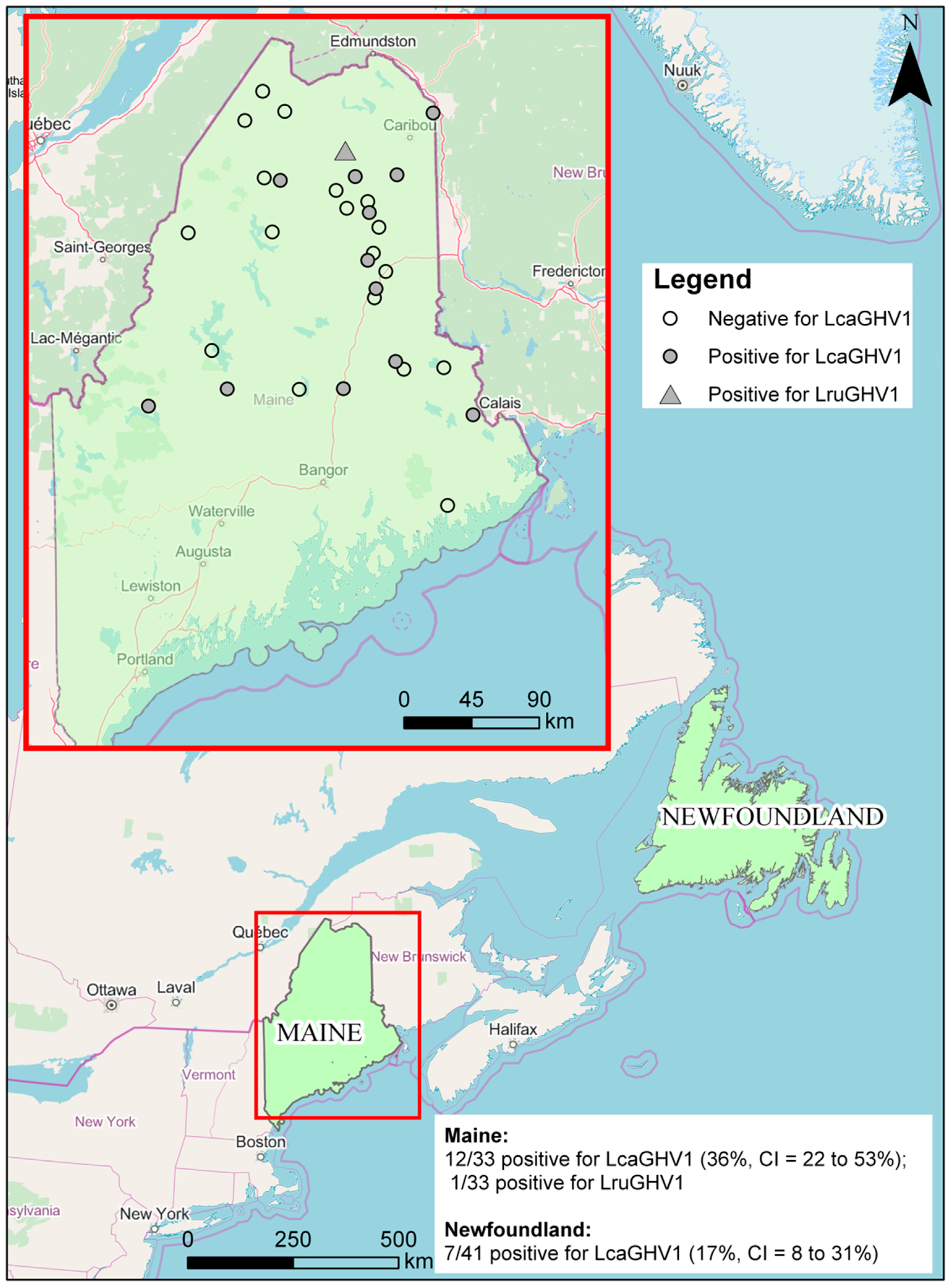
3.2. Prevalence of LcaGHV1 in Lynx from Maine and Newfoundland
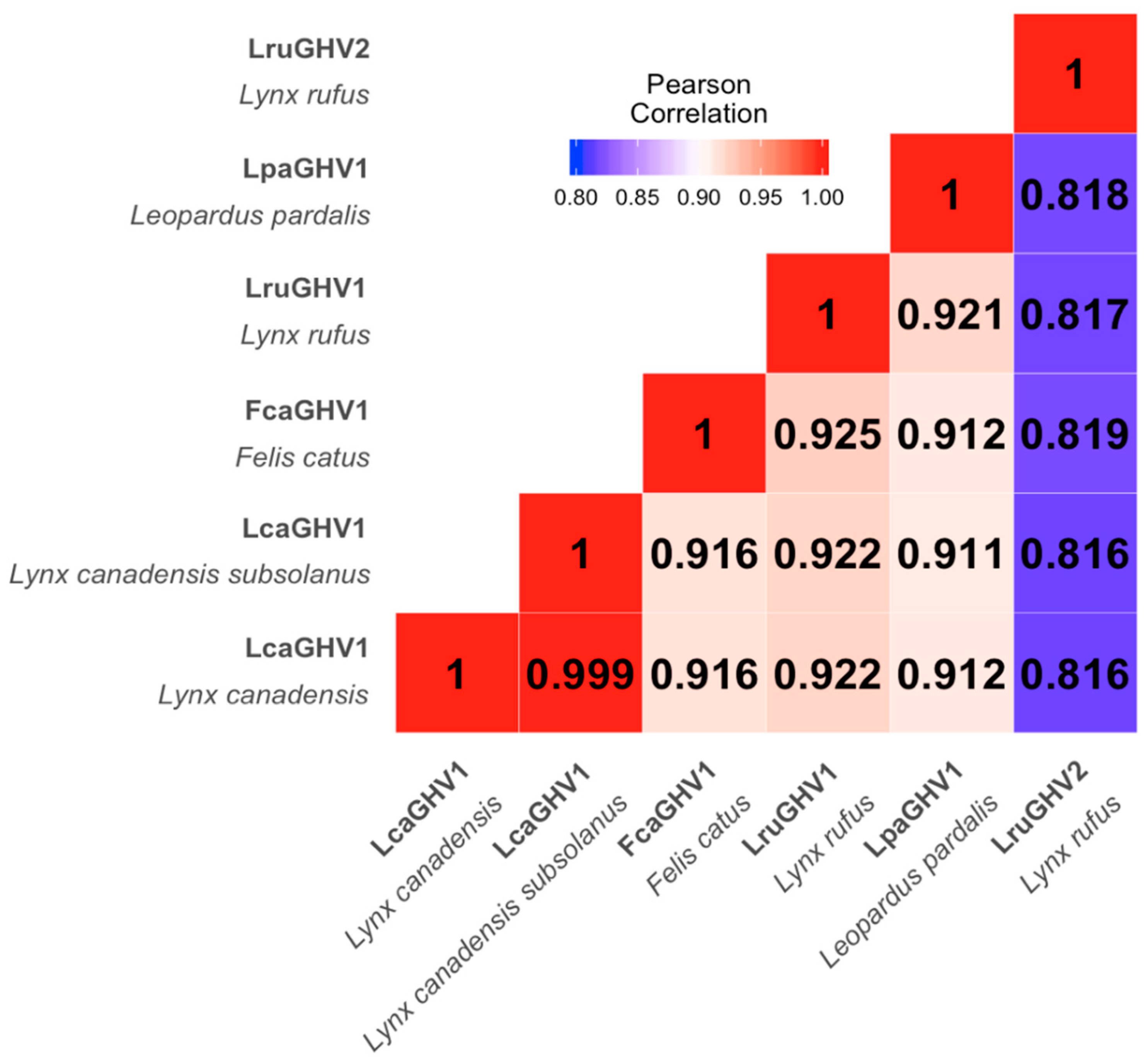
3.3. Sequence Comparison of LcaGHV1 from Maine and Newfoundland
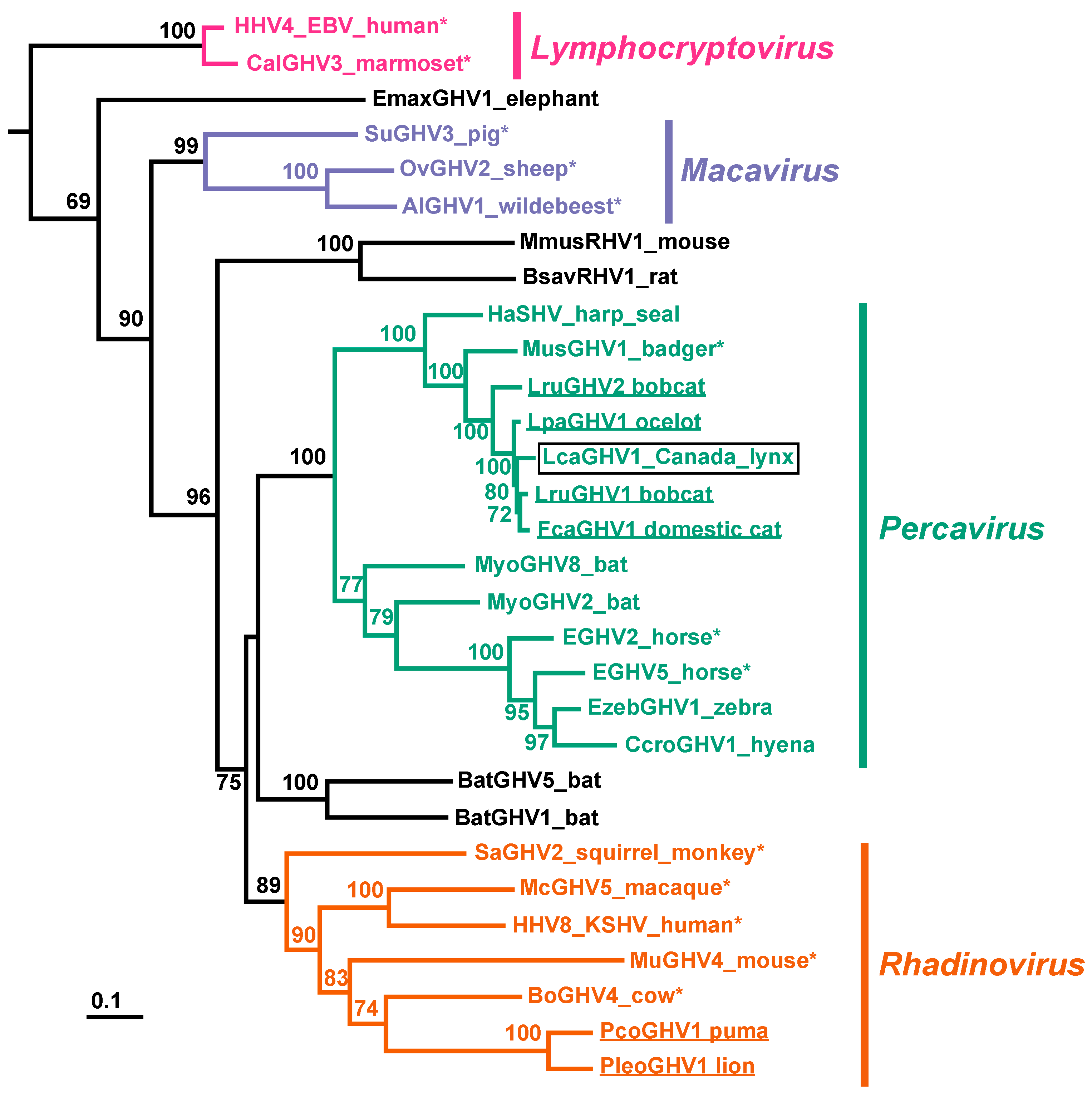
3.4. Phylogenetic Characterization of LcaGHV1
3.5. Risk Factors for LcaGHV1 Infection in Lynx
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- King, A.M.Q.; Adams, M.J.; Carsten, E.B.; Lefkowitz, E.J. Virus Taxonomy: Classification and Nomenclature of Viruses; Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: Oxford, UK, 2012; ISBN 978-0-12-384684-6. [Google Scholar]
- Barton, E.; Mandal, P.; Speck, S.H. Pathogenesis and Host Control of Gammaherpesviruses: Lessons from the Mouse. Annu. Rev. Immunol. 2011, 29, 351–397. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J.; Rixon, F.J.; Davison, A.J. Topics in herpesvirus genomics and evolution. Virus Res. 2006, 117, 90–104. [Google Scholar] [CrossRef]
- Ehlers, B.; Dural, G.; Yasmum, N.; Lembo, T.; de Thoisy, B.; Ryser-Degiorgis, M.-P.; Ulrich, R.G.; McGeoch, D.J. Novel mammalian herpesviruses and lineages within the Gammaherpesvirinae: Cospeciation and interspecies transfer. J. Virol. 2008, 82, 3509–3516. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, B.; Kuchler, J.; Yasmum, N.; Dural, G.; Voigt, S.; Schmidt-Chanasit, J.; Jakel, T.; Matuschka, F.-R.; Richter, D.; Essbauer, S.; et al. Identification of Novel Rodent Herpesviruses, Including the First Gammaherpesvirus of Mus musculus. J. Virol. 2007, 81, 8091–8100. [Google Scholar] [CrossRef] [PubMed]
- Wibbelt, G.; Kurth, A.; Yasmum, N.; Bannert, M.; Nagel, S.; Nitsche, A.; Ehlers, B. Discovery of herpesviruses in bats. J. Gen. Virol. 2007, 88, 2651–2655. [Google Scholar] [CrossRef] [PubMed]
- Chmielewicz, B.; Goltz, M.; Ehlers, B. Detection and multigenic characterization of a novel gammaherpesvirus in goats. Virus Res. 2001, 75, 87–94. [Google Scholar] [CrossRef]
- Chmielewicz, B.; Goltz, M.; Franz, T.; Bauer, C.; Brema, S.; Ellerbrok, H.; Beckmann, S.; Rziha, H.J.; Lahrmann, K.H.; Romero, C.; et al. A novel porcine gammaherpesvirus. Virology 2003, 308, 317–329. [Google Scholar] [CrossRef]
- Cabello, J.; Esperón, F.; Napolitano, C.; Hidalgo, E.; Dávila, J.A.; Millán, J. Molecular identification of a novel gammaherpesvirus in the endangered Darwin’s fox (Lycalopex fulvipes). J. Gen. Virol. 2013, 94, 2745–2749. [Google Scholar] [CrossRef]
- Shabman, R.S.; Shrivastava, S.; Tsibane, T.; Attie, O.; Jayaprakash, A.; Mire, C.E.; Dilley, K.E.; Puri, V.; Stockwell, T.B.; Geisbert, T.W.; et al. Isolation and Characterization of a Novel Gammaherpesvirus from a Microbat Cell Line. mSphere 2016, 1. [Google Scholar] [CrossRef]
- Wu, Z.; Ren, X.; Yang, L.; Hu, Y.; Yang, J.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Du, J.; et al. Virome Analysis for Identification of Novel Mammalian Viruses in Bat Species from Chinese Provinces. J. Virol. 2012, 86, 10999–11012. [Google Scholar] [CrossRef]
- Jung, J.U.; Speck, S.H. Insights into chronic gamma-herpesvirus infections. Curr. Opin. Virol. 2013, 3, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Smith, M.D.; Smith, D.M.; Scheffler, K.; Kosakovsky Pond, S.L. Evolutionary origins of human herpes simplex viruses 1 and 2. Mol. Biol. Evol. 2014, 31, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M. Pathogenesis of gammaherpesvirus infections. Vet. Microbiol. 2006, 113, 211–222. [Google Scholar] [CrossRef]
- Cesarman, E. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett. 2011, 305, 163–174. [Google Scholar] [CrossRef]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Russell, G.C.; Stewart, J.P.; Haig, D.M. Malignant catarrhal fever: A review. Vet. J. 2009, 179, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J.; Robinson, N.E.; Lim, A.; Brandenberger, C.; Maes, R.; Behan, A.; Bolin, S.R. Experimental Induction of Pulmonary Fibrosis in Horses with the Gammaherpesvirus Equine Herpesvirus 5. PLoS ONE 2013, 8, e77754. [Google Scholar] [CrossRef] [PubMed]
- Speck, S.H.; Ganem, D. Viral latency and its regulation: Lessons from the γ-Herpesviruses. Cell Host Microbe 2010, 8, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.J.; Kipar, A.; Sample, J.T.; Stewart, J.P. Pathogenesis of a Model Gammaherpesvirus in a Natural Host. J. Virol. 2010, 84, 3949–3961. [Google Scholar] [CrossRef]
- Beatty, J.A.; Troyer, R.M.; Carver, S.; Barrs, V.R.; Espinasse, F.; Conradi, O.; Stutzman-Rodriguez, K.; Chan, C.C.; Tasker, S.; Lappin, M.R.; et al. Felis catus gammaherpesvirus 1; a widely endemic potential pathogen of domestic cats. Virology 2014, 460–461, 100–107. [Google Scholar] [CrossRef]
- Troyer, R.M.; Beatty, J.A.; Stutzman-Rodriguez, K.R.; Carver, S.; Lozano, C.C.; Lee, J.S.; Lappin, M.R.; Riley, S.P.D.; Serieys, L.E.K.; Logan, K.A.; et al. Novel Gammaherpesviruses in North American Domestic Cats, Bobcats, and Pumas: Identification, Prevalence, and Risk Factors. J. Virol. 2014, 88, 3914–3924. [Google Scholar] [CrossRef] [PubMed]
- Ertl, R.; Korb, M.; Langbein-Detsch, I.; Klein, D. Prevalence and risk factors of gammaherpesvirus infection in domestic cats in Central Europe. Virol. J. 2015, 12, 146. [Google Scholar] [CrossRef]
- Tateno, M.; Takahashi, M.; Miyake, E.; Nishigaki, K.; Tsujimoto, H.; Endo, Y. Molecular epidemiological study of gammaherpesvirus in domestic cats in Japan. J. Vet. Med. Sci. 2017, 79, 1735–1740. [Google Scholar] [CrossRef] [PubMed]
- Kurissio, J.K.; Rodrigues, M.V.; Taniwaki, S.A.; de Zanutto, M.S.; Filoni, C.; Galdino, M.V.; Araújo Júnior, J.P. Felis catus gammaherpesvirus 1 (FcaGHV1) and coinfections with feline viral pathogens in domestic cats in Brazil. Ciência Rural 2018, 48. [Google Scholar] [CrossRef]
- Lozano, C.C.; Sweanor, L.L.; Wilson-Henjum, G.; Kays, R.W.; Moreno, R.; VandeWoude, S.; Troyer, R.M. Identification of novel gammaherpesviruses in ocelots (Leopardus pardalis) and bobcats (Lynx rufus). J. Wildl. Dis. 2015, 51, 911–915. [Google Scholar] [CrossRef]
- Canuti, M.; Rodrigues, B.; Whitney, H.G.; Lang, A.S. Introduction of canine parvovirus 2 into wildlife on the Island of Newfoundland, Canada. Infect. Genet. Evol. 2017, 55, 205–208. [Google Scholar] [CrossRef]
- Koen, E.L.; Bowman, J.; Wilson, P.J. Isolation of peripheral populations of Canada lynx (Lynx canadensis). Can. J. Zool. 2015, 93, 521–530. [Google Scholar] [CrossRef]
- Van Zyll De Jong, C.G. Differentiation of the Canada lynx, Felis (Lynx) canadensis subsolana, in Newfoundland. Can. J. Zool. 1975, 53, 699–705. [Google Scholar] [CrossRef]
- Row, J.R.; Gomez, C.; Koen, E.L.; Bowman, J.; Murray, D.L.; Wilson, P.J. Dispersal promotes high gene flow among Canada lynx populations across mainland North America. Conserv. Genet. 2012, 13, 1259–1268. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Rose, T.M.; Strand, K.B.; Schultz, E.R.; Schaefer, G.; Rankin, G.W.; Thouless, M.E.; Tsai, C.C.; Bosch, M.L. Identification of two homologs of the Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in retroperitoneal fibromatosis of different macaque species. J. Virol. 1997, 71, 4138–4144. [Google Scholar] [PubMed]
- Team, R.C. R: A Language and Environment for Statistical Computing. Available online: https://www.gbif.org/tool/81287/r-a-language-and-environment-for-statistical-computing (accessed on 18 October 2018).
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Delport, W.; Poon, A.F.Y.; Frost, S.D.W.; Kosakovsky Pond, S.L. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef]
- Delport, W.; Scheffler, K.; Botha, G.; Gravenor, M.B.; Muse, S.V.; Kosakovsky Pond, S.L.K. CodonTest: Modeling amino acid substitution preferences in coding sequences. PLoS Comput. Biol. 2010, 6, e1000885. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef]
- Loisel, D.A.; Troyer, R.M.; VandeWoude, S. High prevalence of Lynx rufus gammaherpesvirus 1 in wild Vermont bobcats. PeerJ 2018, 6, e4982. [Google Scholar] [CrossRef] [PubMed]
- McLuckie, A.; Tasker, S.; Dhand, N.K.; Spencer, S.; Beatty, J.A. High prevalence of Felis catus gammaherpesvirus 1 infection in haemoplasma-infected cats supports co-transmission. Vet. J. 2016, 214, 117–121. [Google Scholar] [CrossRef]
- Stutzman-Rodriguez, K.; Rovnak, J.; VandeWoude, S.; Troyer, R.M. Domestic cats seropositive for Felis catus gammaherpesvirus 1 are often qPCR negative. Virology 2016, 498, 23–30. [Google Scholar] [CrossRef]
- Gehrt, S.D.; Riley, S.P.D.; Cypher, B.L. Urban Carnivores: Ecology, Conflict, and Conservation; JHU Press: Baltimore, MD, USA, 2010; ISBN 0801893895. [Google Scholar]
- Clark, J.R. Determination of Threatened Status for the Contiguous U.S. Distinct Population Segment of the Canada Lynx and Related Rule; Final Rule. Fed. Regist. 2000, 65, 16052–16086. [Google Scholar]
- Vashon, J.H.; Meehan, A.L.; Jakubas, W.J.; Organ, J.F.; Vashon, A.D.; McLaughlin, C.R.; Matula, G.J.; Crowley, S.M. Spatial Ecology of a Canada Lynx Population in Northern Maine. J. Wildl. Manag. 2008, 72, 1479–1487. [Google Scholar] [CrossRef]
- Peers, M.J.L.; Thornton, D.H.; Murray, D.L. Evidence for large-scale effects of competition: Niche displacement in canada lynx and bobcat. Proc. R. Soc. B Biol. Sci. 2013, 280, 20132495. [Google Scholar] [CrossRef] [PubMed]
- Homyack, J.A.; Vashon, J.H.; Libby, C.; Lindquist, E.L.; Loch, S.; McAlpine, D.F. Canada Lynx-bobcat (Lynx canadensis × L. rufus) Hybrids at the Southern Periphery of Lynx range in Maine, Minnesota and New Brunswick. Am. Midl. Nat. 2008, 159, 504–509. [Google Scholar] [CrossRef]
- Makundi, I.; Koshida, Y.; Endo, Y.; Nishigaki, K. Identification of Felis catus Gammaherpesvirus 1 in Tsushima Leopard Cats (Prionailurus bengalensis euptilurus) on Tsushima Island, Japan. Viruses 2018, 10, 378. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Eizirik, E.; Pecon-Slattery, J.; Murphy, W.J.; Antunes, A.; Teeling, E.; O’Brien, S.J. The late miocene radiation of modern felidae: A genetic assesstment. Science 2006, 311, 73–77. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Set | Primer Name | Use 1 | Sequence (5’ → 3’) |
|---|---|---|---|
| Carnivore GAPDH | GAPcons-F | for | CATCAAGAAGGTGGTGAAGCA |
| GAPcons-R | rev | GGAAATGAGCTTGACAAAGTGG | |
| LcaGHV1 gB | gBLynx-F1 | 1st for | TATCTATGGAAAGCCAGTTTCAG |
| gBLynx-R1 | 1st rev | ACCACCTCAAAGTCAATGTTCT | |
| gBLynx-F4 | 2nd for | GAGTGTGTCATTGTAGACCAAACCAAG | |
| gBLynx-R4 | 2nd rev | TTGTGCATTTGATGCCCTGAC | |
| LcaGHV1 long | polLynx-R1 | 1st rev | CGTTATATGCACAGTTGAGTGG |
| polLynx-R2 | 2nd rev | ACAGGGGTGAGGGATTCA |
| Degenerate PCR Protocol | Tissue Type | |||
|---|---|---|---|---|
| Spleen | Jejunum | Lung | Bone Marrow | |
| Pan-GHV gB | 1/5 (17–4019 1) | 0/5 | 0/3 | - |
| Pan-HV DNAPol | 0/5 | 0/5 | 0/3 | 0/3 |
| Percavirus DNApol | 1/5 (17–4019 1) | 1/5 (17–4019 1) | 0/3 | 0/3 |
| No. Samples | ||||
|---|---|---|---|---|
| Predictor | LcaGHV1-Negative | LcaGHV1-Positive | % Positive | p-Value |
| Sex 1 | 0.854 | |||
| Male | 11 | 7 | 39.0 | |
| Female | 9 | 5 | 35.7 | |
| Unknown | 1 | 0 | 0.0 | |
| Location | 0.064 | |||
| Newfoundland | 34 | 7 | 17.1 | |
| Maine | 21 | 12 | 36.4 | |
| Lungworms 1 | 0.919 | |||
| Positive | 13 | 8 | 38.1 | |
| Negative | 6 | 4 | 40.0 | |
| Lung Inflammation 1 | 0.381 | |||
| Present | 11 | 5 | 31.3 | |
| Absent | 8 | 7 | 46.7 | |
| Heart and Muscle Inflammation 1 | 0.394 | |||
| Present | 12 | 5 | 29.4 | |
| Absent | 9 | 7 | 43.8 | |
| Weight 1 | 0.296 | |||
| Mean | 8.06 | 9.05 | ||
| n | 16 | 9 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hendrikse, L.D.; Kambli, A.; Kayko, C.; Canuti, M.; Rodrigues, B.; Stevens, B.; Vashon, J.; Lang, A.S.; Needle, D.B.; Troyer, R.M. Identification of a Novel Gammaherpesvirus in Canada lynx (Lynx canadensis). Viruses 2019, 11, 363. https://doi.org/10.3390/v11040363
Hendrikse LD, Kambli A, Kayko C, Canuti M, Rodrigues B, Stevens B, Vashon J, Lang AS, Needle DB, Troyer RM. Identification of a Novel Gammaherpesvirus in Canada lynx (Lynx canadensis). Viruses. 2019; 11(4):363. https://doi.org/10.3390/v11040363
Chicago/Turabian StyleHendrikse, Liam D., Ankita Kambli, Caroline Kayko, Marta Canuti, Bruce Rodrigues, Brian Stevens, Jennifer Vashon, Andrew S. Lang, David B. Needle, and Ryan M. Troyer. 2019. "Identification of a Novel Gammaherpesvirus in Canada lynx (Lynx canadensis)" Viruses 11, no. 4: 363. https://doi.org/10.3390/v11040363
APA StyleHendrikse, L. D., Kambli, A., Kayko, C., Canuti, M., Rodrigues, B., Stevens, B., Vashon, J., Lang, A. S., Needle, D. B., & Troyer, R. M. (2019). Identification of a Novel Gammaherpesvirus in Canada lynx (Lynx canadensis). Viruses, 11(4), 363. https://doi.org/10.3390/v11040363