Discovery and Characterization of Novel Bat Coronavirus Lineages from Kazakhstan

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
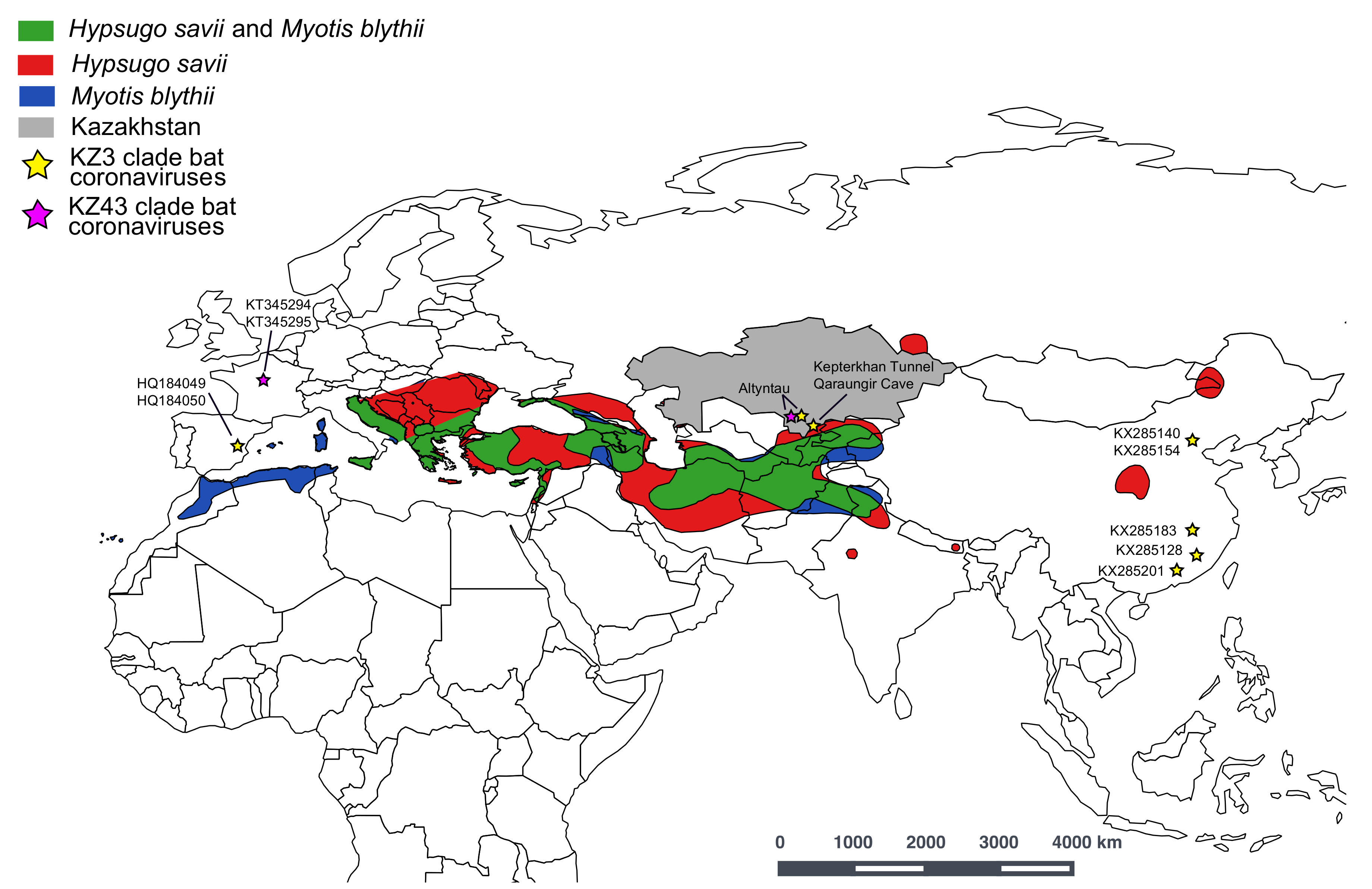
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Hutson, A.M.; Mickleburgh, S.P. Microchiropteran Bats: Global Status Survey And Conservation Action Plan. IUCN, 2001; Volume 56. Available online: http://www.iucn.org/dbtw-wpd/edocs/2001-008.pdf (accessed on 29 January 2019).
- Kunz, T.H.; Fenton, M.B. Bat Ecology; University of Chicago Press: Chicago, IL, USA, 2005. [Google Scholar]
- Hayman, D.T. Bats as viral reservoirs. Annu. Rev. Virol. 2016, 3, 77–99. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.; Wang, L.F. Bats and their virome: An important source of emerging viruses capable of infecting humans. Curr. Opin. Virol. 2013, 3, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef]
- Vijaykrishna, D.; Smith, G.J.; Zhang, J.X.; Peiris, J.S.; Chen, H.; Guan, Y. Evolutionary insights into the ecology of coronaviruses. J. Virol. 2007, 81, 4012–4020. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoek, L. Human coronaviruses: What do they cause? Antivir. Ther. 2007, 12, 651. [Google Scholar] [PubMed]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Ithete, N.L.; Richards, L.R.; Schoeman, M.C.; Preiser, W.; Drosten, C.; Drexler, J.F. Rooting the phylogenetic tree of middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat. J. Virol. 2014, 88, 11297–11303. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.-L.; Shi, W.-F.; Zhang, W.; Zhu, Y.; Zhang, Y.-W.; Xie, Q.-M.; Mani, S.; et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Gromov, I.M.; Baranova, G.; Baryshnikov, G. Catalog of the Mammals of USSR; Nauka: Alma-Ata, Kazakhstan, 1981; Volume 1, p. 456. [Google Scholar]
- Bekenov, A.; Butovskyi, P.; Kasabekov, B.; Lankin, P.; Strelkov, P.; Stogov, I. Mammals of Kazakhstan; Nauka: Alma-Ata, Kazakhstan, 1985; Volume 4. [Google Scholar]
- Jargalsaikhan, A. Bat study in the Kharaa region, Mongolia. J. Asia-Pac. Biodivers. 2016, 9, 107–115. [Google Scholar] [CrossRef][Green Version]
- Tiunov, M. Bats of the Russian Far East. Dalnauka Vladivostok 1997, 134. [Google Scholar]
- Watanabe, S.; Masangkay, J.S.; Nagata, N.; Morikawa, S.; Mizutani, T.; Fukushi, S.; Alviola, P.; Omatsu, T.; Ueda, N.; Iha, K.; et al. Bat coronaviruses and experimental infection of bats, the Philippines. Emerg. Infect. Dis. 2010, 16, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Bininda-Emonds, O.R. transAlign: Using amino acids to facilitate the multiple alignment of protein-coding DNA sequences. BMC Bioinformatics 2005, 6, 156. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Bilgin, İ.R. The conservation genetics of three cave-dwelling bat species in southeastern Europe and Anatolia. Turk. J. Zool. 2012, 36, 275–282. [Google Scholar]
- Peel, A.J.; Sargan, D.R.; Baker, K.S.; Hayman, D.T.; Barr, J.A.; Crameri, G.; Suu-Ire, R.; Broder, C.C.; Lembo, T.; Wang, L.-F. Continent-wide panmixia of an African fruit bat facilitates transmission of potentially zoonotic viruses. Nature Commun. 2013, 4, 2770. [Google Scholar] [CrossRef] [PubMed]
- Juste, J.P.; Paunović, M. Hypsugo savii. The IUCN Red List of Threatened Species 2016: E.T44856A22072380. Available online: https://www.iucnredlist.org/species/44856/22072380 (accessed on 29 January 2019).
- Juste, J.P.; Paunović, M. Myotis blythii. The IUCN Red List of Threatened Species 2016: E.T14124A22053297. Available online: https://www.iucnredlist.org/species/14124/22053297 (accessed on on 29 January 2019).
- Hutterer, R.; Ivanova, T.; Meyers-Cords, C.H.; Rodrigues, L. Bat migrations in Europe: A review of banding data and literature; Federal Agency for Nature Conservation: Bonn, Germany, 2005; Volume 28. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
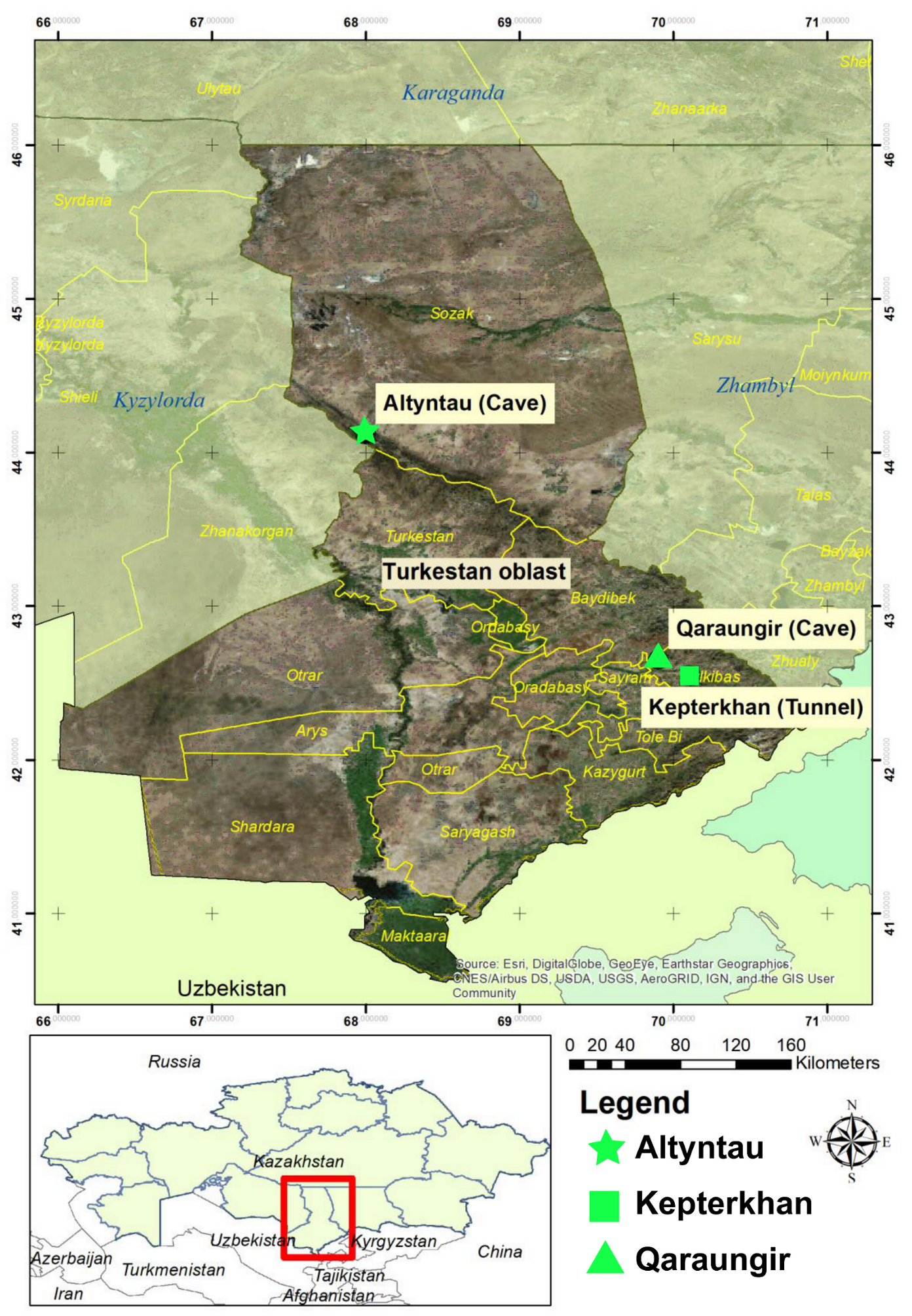
| Sample Collection Site | GIS Coordinates | Samples Tested | Positives (%) |
|---|---|---|---|
| Kepterkhan tunnel, Tulkibas rayon, Turkestan oblast | N 42°32′ 36.5″ E 70°06′ 46.9″ | 101 | 7 (6.9%) |
| Qaraungir cave, Tulkibas rayon, Turkestan oblast | N 42°39′ 04.7″ E 69°54′ 02.6″ | 50 | 12 (24%) |
| Ungirli cave, Altyntau, Sozak rayon, Turkestan oblast | N 44°06′ 59.4″ E 67° 59′ 47.9″ | 49 | 6 (12.2%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendenhall, I.H.; Kerimbayev, A.A.; Strochkov, V.M.; Sultankulova, K.T.; Kopeyev, S.K.; Su, Y.C.F.; Smith, G.J.D.; Orynbayev, M.B. Discovery and Characterization of Novel Bat Coronavirus Lineages from Kazakhstan. Viruses 2019, 11, 356. https://doi.org/10.3390/v11040356
Mendenhall IH, Kerimbayev AA, Strochkov VM, Sultankulova KT, Kopeyev SK, Su YCF, Smith GJD, Orynbayev MB. Discovery and Characterization of Novel Bat Coronavirus Lineages from Kazakhstan. Viruses. 2019; 11(4):356. https://doi.org/10.3390/v11040356
Chicago/Turabian StyleMendenhall, Ian H., Aslan A. Kerimbayev, Vitaliy M. Strochkov, Kulyaisan T. Sultankulova, Syrym K. Kopeyev, Yvonne C.F. Su, Gavin J.D. Smith, and Mukhit B. Orynbayev. 2019. "Discovery and Characterization of Novel Bat Coronavirus Lineages from Kazakhstan" Viruses 11, no. 4: 356. https://doi.org/10.3390/v11040356
APA StyleMendenhall, I. H., Kerimbayev, A. A., Strochkov, V. M., Sultankulova, K. T., Kopeyev, S. K., Su, Y. C. F., Smith, G. J. D., & Orynbayev, M. B. (2019). Discovery and Characterization of Novel Bat Coronavirus Lineages from Kazakhstan. Viruses, 11(4), 356. https://doi.org/10.3390/v11040356