Whole-Genome Analysis of Human Papillomavirus Type 16 Prevalent in Japanese Women with or without Cervical Lesions

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Samples
2.2. Viral Whole-Genome Amplification and Next Generation Sequencing
2.3. Phylogenetic Tree Construction
2.4. Sequencing of HPV16 E6/E7
2.5. Generation of Human Cervical Keratinocytes Expressing E7
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
3.1. Determination of HPV16 Whole-Genome Sequences from Japanese Women
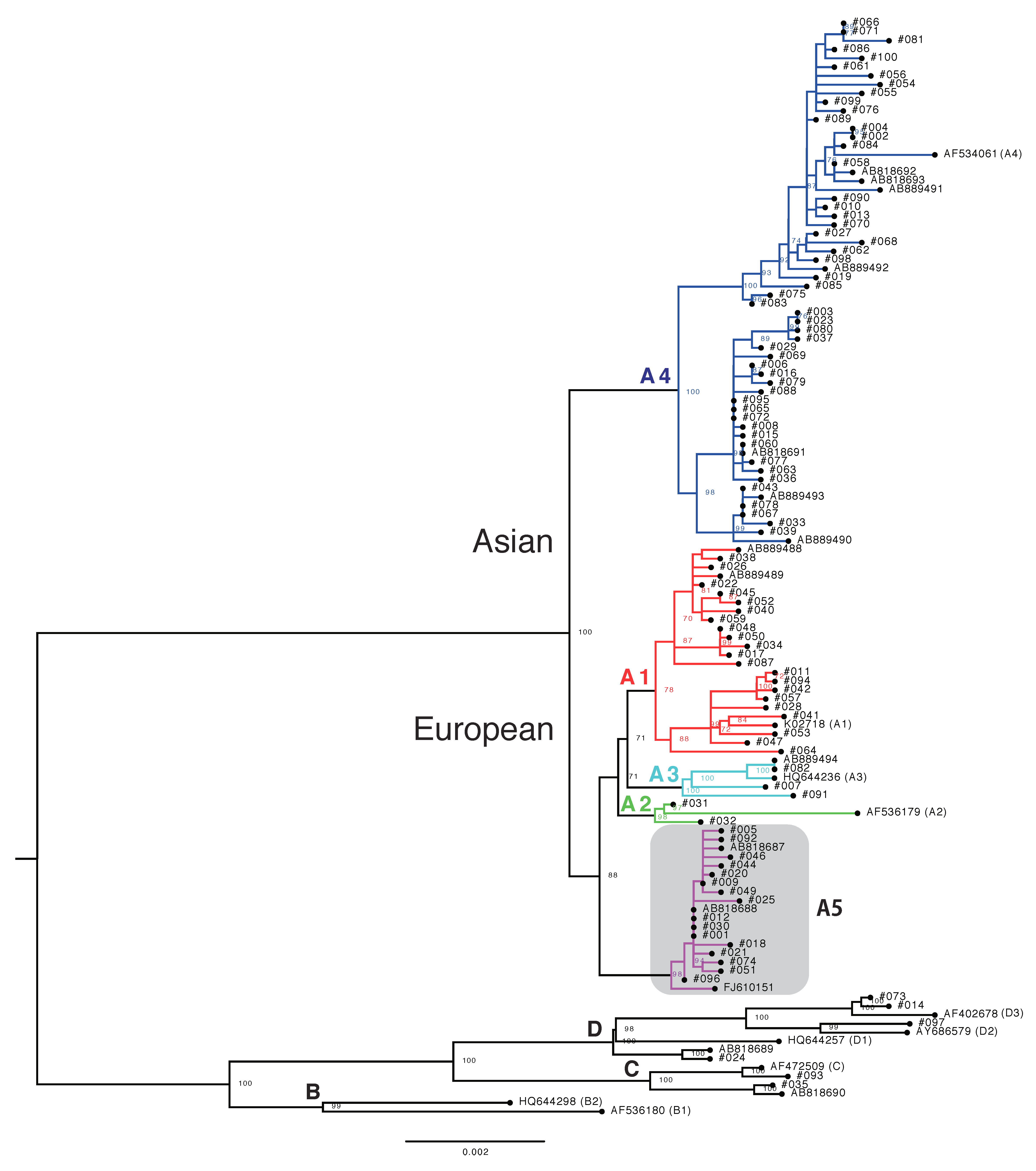
3.2. Phylogenetic Analysis of Japanese HPV16 Isolates
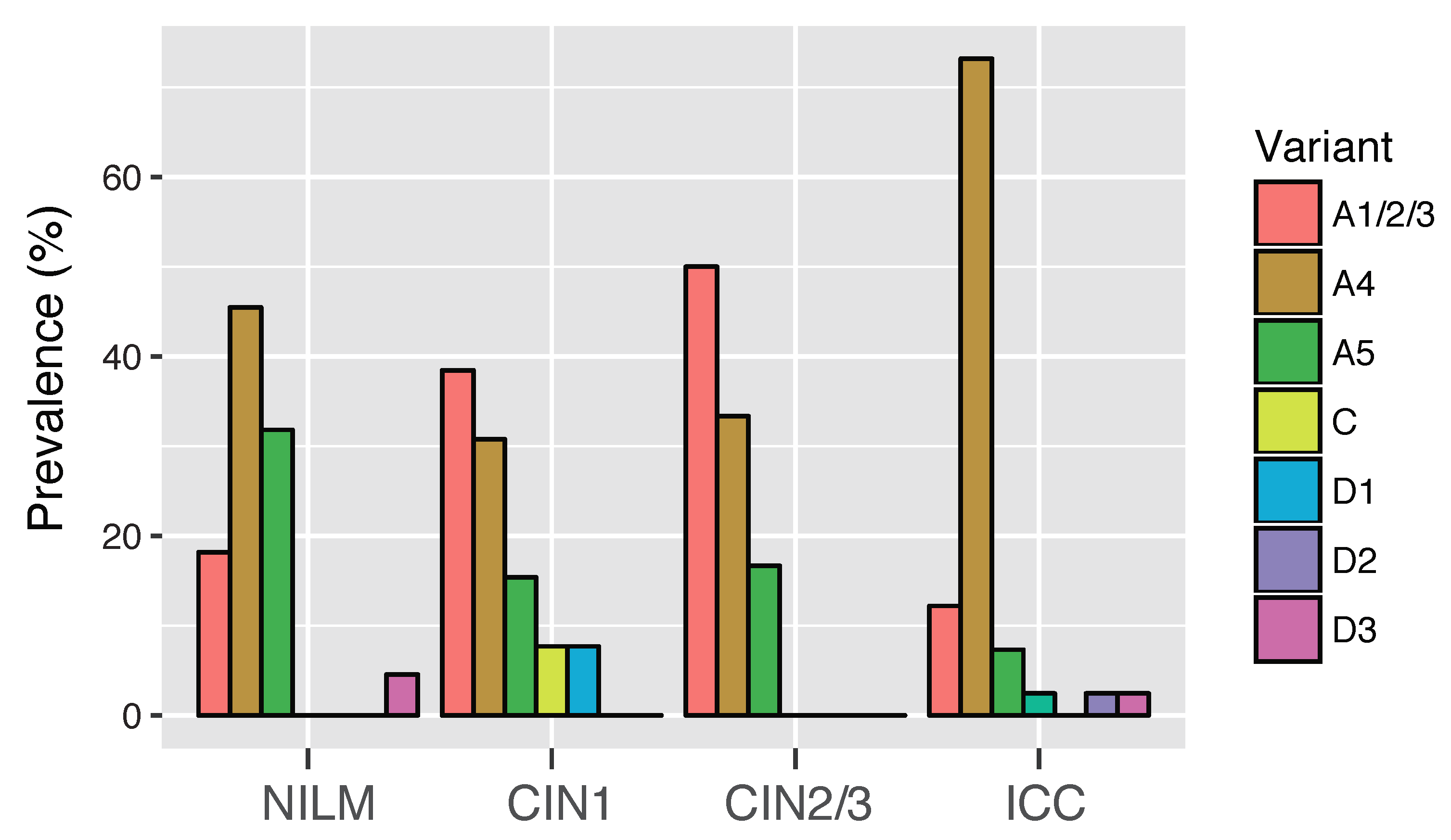
3.3. Risk Assessment of HPV16 Variants for Cervical Cancer Development
3.4. Genetic Variability among Sublineage A4 Variants
3.5. Identification of SNPs Unique for A5 Variants
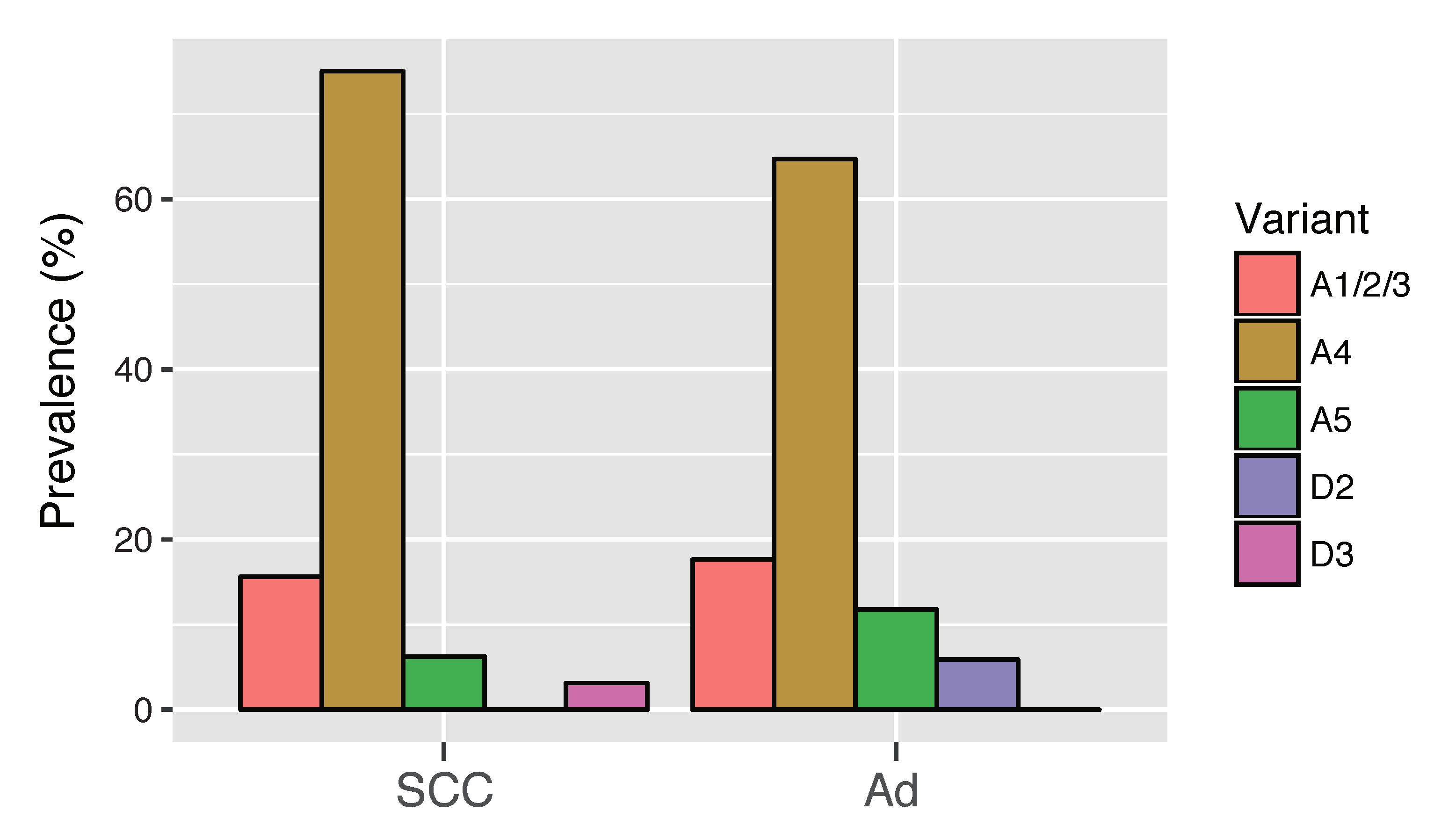
3.6. HPV16 Variant Distribution in Cervical Adenocarcinoma
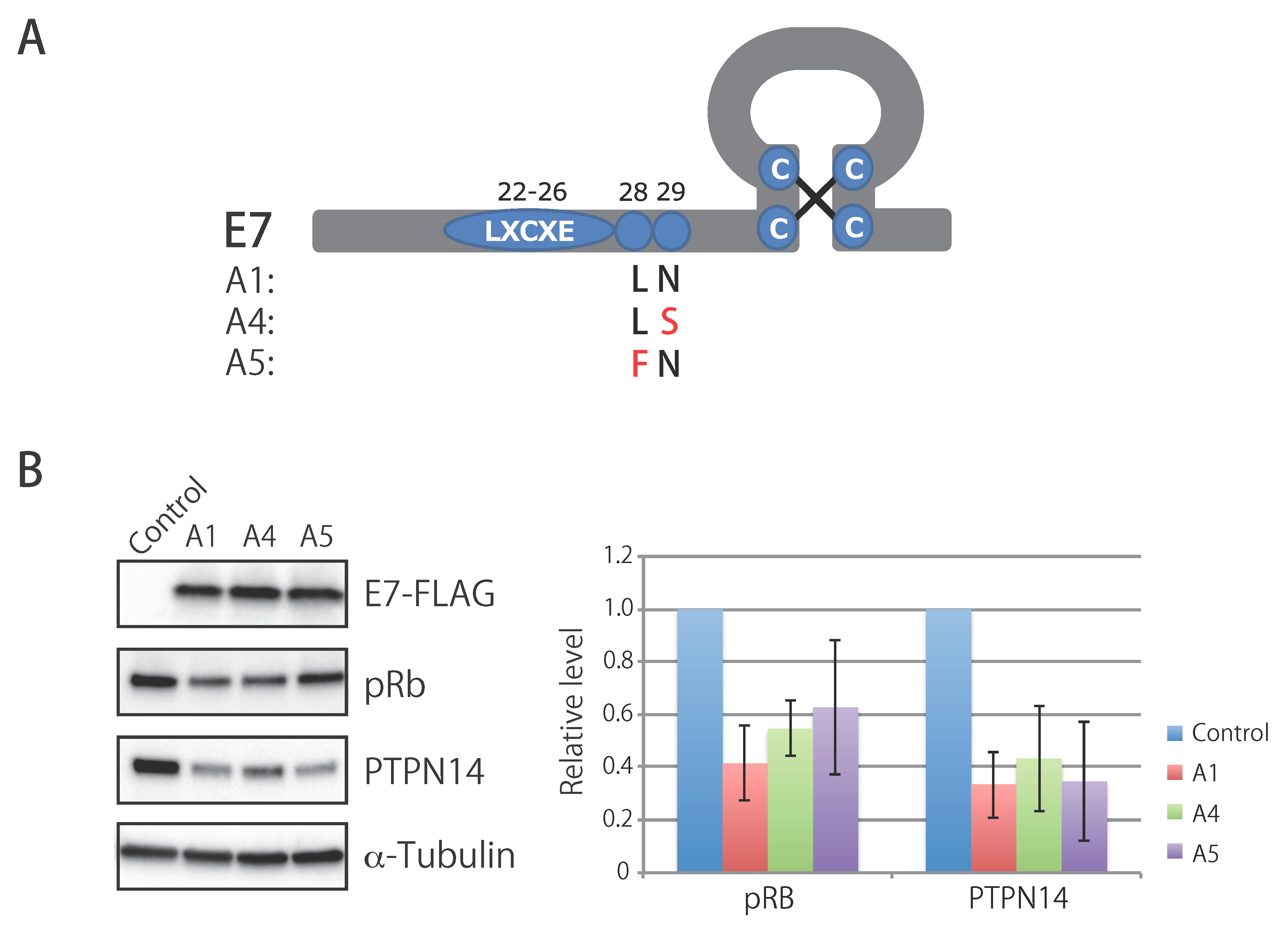
3.7. Biological Activity of E7 Variants of Lineage A
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- de Sanjose, S.; Quint, W.G.; Alemany, L.; Geraets, D.T.; Klaustermeier, J.E.; Lloveras, B.; Tous, S.; Felix, A.; Bravo, L.E.; Shin, H.R.; et al. Human papillomavirus genotype attribution in invasive cervical cancer: A retrospective cross-sectional worldwide study. Lancet Oncol. 2010, 11, 1048–1056. [Google Scholar] [CrossRef]
- Giuliano, A.R.; Tortolero-Luna, G.; Ferrer, E.; Burchell, A.N.; de Sanjose, S.; Kjaer, S.K.; Munoz, N.; Schiffman, M.; Bosch, F.X. Epidemiology of human papillomavirus infection in men, cancers other than cervical and benign conditions. Vaccine 2008, 26 (Suppl. 10), K17–K28. [Google Scholar] [CrossRef]
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.D.; Harari, A.; Chen, Z. Human papillomavirus genome variants. Virology 2013, 445, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wakae, K.; Kitamura, K.; Aoyama, S.; Liu, G.; Koura, M.; Monjurul, A.M.; Kukimoto, I.; Muramatsu, M. APOBEC3 deaminases induce hypermutation in human papillomavirus 16 DNA upon beta interferon stimulation. J. Virol. 2014, 88, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Onuki, M.; Tenjimbayashi, Y.; Mori, S.; Ishii, Y.; Takeuchi, T.; Tasaka, N.; Satoh, T.; Morisada, T.; Iwata, T.; et al. Within-host variations of human papillomavirus reveal APOBEC signature mutagenesis in the viral genome. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Van Doorslaer, K.; Pandey, A.; Espinosa, J.M.; Pyeon, D. Role of the host restriction factor APOBEC3 on papillomavirus evolution. Virus Evol. 2015, 1, vev015. [Google Scholar] [CrossRef] [PubMed]
- Hildesheim, A.; Schiffman, M.; Bromley, C.; Wacholder, S.; Herrero, R.; Rodriguez, A.; Bratti, M.C.; Sherman, M.E.; Scarpidis, U.; Lin, Q.Q.; et al. Human papillomavirus type 16 variants and risk of cervical cancer. J. Natl. Cancer Inst. 2001, 93, 315–318. [Google Scholar] [CrossRef]
- Sichero, L.; Ferreira, S.; Trottier, H.; Duarte-Franco, E.; Ferenczy, A.; Franco, E.L.; Villa, L.L. High grade cervical lesions are caused preferentially by non-European variants of HPVs 16 and 18. Int. J. Cancer 2007, 120, 1763–1768. [Google Scholar] [CrossRef]
- Xi, L.F.; Koutsky, L.A.; Hildesheim, A.; Galloway, D.A.; Wheeler, C.M.; Winer, R.L.; Ho, J.; Kiviat, N.B. Risk for high-grade cervical intraepithelial neoplasia associated with variants of human papillomavirus types 16 and 18. Cancer Epidemiol. Biomark. Prev. 2007, 16, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Rodriguez, A.C.; Chen, Z.; Wacholder, S.; Herrero, R.; Hildesheim, A.; Desalle, R.; Befano, B.; Yu, K.; Safaeian, M.; et al. A population-based prospective study of carcinogenic human papillomavirus variant lineages, viral persistence, and cervical neoplasia. Cancer Res. 2010, 70, 3159–3169. [Google Scholar] [CrossRef]
- Freitas, L.B.; Chen, Z.; Muqui, E.F.; Boldrini, N.A.; Miranda, A.E.; Spano, L.C.; Burk, R.D. Human papillomavirus 16 non-European variants are preferentially associated with high-grade cervical lesions. PLoS ONE 2014, 9, e100746. [Google Scholar] [CrossRef]
- Berumen, J.; Ordonez, R.M.; Lazcano, E.; Salmeron, J.; Galvan, S.C.; Estrada, R.A.; Yunes, E.; Garcia-Carranca, A.; Gonzalez-Lira, G.; Madrigal-de la Campa, A. Asian-American variants of human papillomavirus 16 and risk for cervical cancer: A case-control study. J. Natl. Cancer Inst. 2001, 93, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.D.; Terai, M.; Gravitt, P.E.; Brinton, L.A.; Kurman, R.J.; Barnes, W.A.; Greenberg, M.D.; Hadjimichael, O.C.; Fu, L.; McGowan, L.; et al. Distribution of human papillomavirus types 16 and 18 variants in squamous cell carcinomas and adenocarcinomas of the cervix. Cancer Res. 2003, 63, 7215–7220. [Google Scholar]
- Smith, B.; Chen, Z.; Reimers, L.; van Doorslaer, K.; Schiffman, M.; Desalle, R.; Herrero, R.; Yu, K.; Wacholder, S.; Wang, T.; et al. Sequence imputation of HPV16 genomes for genetic association studies. PLoS ONE 2011, 6, e21375. [Google Scholar] [CrossRef]
- Perez, S.; Cid, A.; Inarrea, A.; Pato, M.; Lamas, M.J.; Couso, B.; Gil, M.; Alvarez, M.J.; Rey, S.; Lopez-Miragaya, I.; et al. Prevalence of HPV 16 and HPV 18 lineages in Galicia, Spain. PLoS ONE 2014, 9, e104678. [Google Scholar] [CrossRef]
- Nicolas-Parraga, S.; Alemany, L.; de Sanjose, S.; Bosch, F.X.; Bravo, I.G.; RIS HPV TT and HPV VVAP Study Groups. Differential HPV16 variant distribution in squamous cell carcinoma, adenocarcinoma and adenosquamous cell carcinoma. Int. J. Cancer 2017, 140, 2092–2100. [Google Scholar] [CrossRef]
- Mirabello, L.; Yeager, M.; Cullen, M.; Boland, J.F.; Chen, Z.; Wentzensen, N.; Zhang, X.; Yu, K.; Yang, Q.; Mitchell, J.; et al. HPV16 sublineage associations with histology-specific cancer risk using HPV whole-genome sequences in 3200 women. J. Natl. Cancer Inst. 2016, 108, djw100. [Google Scholar] [CrossRef] [PubMed]
- Cornet, I.; Gheit, T.; Franceschi, S.; Vignat, J.; Burk, R.D.; Sylla, B.S.; Tommasino, M.; Clifford, G.M. Human papillomavirus type 16 genetic variants: Phylogeny and classification based on E6 and LCR. J. Virol. 2012, 86, 6855–6861. [Google Scholar] [CrossRef]
- Cornet, I.; Gheit, T.; Iannacone, M.R.; Vignat, J.; Sylla, B.S.; Del Mistro, A.; Franceschi, S.; Tommasino, M.; Clifford, G.M. HPV16 genetic variation and the development of cervical cancer worldwide. Br. J. Cancer 2013, 108, 240–244. [Google Scholar] [CrossRef]
- Xi, L.F.; Kiviat, N.B.; Hildesheim, A.; Galloway, D.A.; Wheeler, C.M.; Ho, J.; Koutsky, L.A. Human papillomavirus type 16 and 18 variants: Race-related distribution and persistence. J. Natl. Cancer Inst. 2006, 98, 1045–1052. [Google Scholar] [CrossRef]
- Matsumoto, K.; Yoshikawa, H.; Nakagawa, S.; Tang, X.; Yasugi, T.; Kawana, K.; Sekiya, S.; Hirai, Y.; Kukimoto, I.; Kanda, T.; et al. Enhanced oncogenicity of human papillomavirus type 16 (HPV16) variants in Japanese population. Cancer Lett. 2000, 156, 159–165. [Google Scholar] [CrossRef]
- Hang, D.; Yin, Y.; Han, J.; Jiang, J.; Ma, H.; Xie, S.; Feng, X.; Zhang, K.; Hu, Z.; Shen, H.; et al. Analysis of human papillomavirus 16 variants and risk for cervical cancer in Chinese population. Virology 2016, 488, 156–161. [Google Scholar] [CrossRef]
- Sun, M.; Gao, L.; Liu, Y.; Zhao, Y.; Wang, X.; Pan, Y.; Ning, T.; Cai, H.; Yang, H.; Zhai, W.; et al. Whole genome sequencing and evolutionary analysis of human papillomavirus type 16 in central China. PLoS ONE 2012, 7, e36577. [Google Scholar] [CrossRef]
- Yamada, T.; Manos, M.M.; Peto, J.; Greer, C.E.; Munoz, N.; Bosch, F.X.; Wheeler, C.M. Human papillomavirus type 16 sequence variation in cervical cancers: A worldwide perspective. J. Virol. 1997, 71, 2463–2472. [Google Scholar]
- Sun, Z.; Lu, Z.; Liu, J.; Wang, G.; Zhou, W.; Yang, L.; Liu, C.; Wang, B.; Ruan, Q. Genetic variations of E6 and long control region of human papillomavirus type 16 from patients with cervical lesion in Liaoning, China. BMC Cancer 2013, 13, 459. [Google Scholar] [CrossRef]
- Liu, Y.; Pan, Y.; Gao, W.; Ke, Y.; Lu, Z. Whole-Genome Analysis of Human Papillomavirus Types 16, 18, and 58 Isolated from Cervical Precancer and Cancer Samples in Chinese Women. Sci. Rep. 2017, 7, 263. [Google Scholar] [CrossRef]
- Azuma, Y.; Kusumoto-Matsuo, R.; Takeuchi, F.; Uenoyama, A.; Kondo, K.; Tsunoda, H.; Nagasaka, K.; Kawana, K.; Morisada, T.; Iwata, T.; et al. Human papillomavirus genotype distribution in cervical intraepithelial neoplasia grade 2/3 and invasive cervical cancer in Japanese women. Jpn. J. Clin. Oncol. 2014, 44, 910–917. [Google Scholar] [CrossRef]
- Kukimoto, I.; Maehama, T.; Sekizuka, T.; Ogasawara, Y.; Kondo, K.; Kusumoto-Matsuo, R.; Mori, S.; Ishii, Y.; Takeuchi, T.; Yamaji, T.; et al. Genetic variation of human papillomavirus type 16 in individual clinical specimens revealed by deep sequencing. PLoS ONE 2013, 8, e80583. [Google Scholar] [CrossRef]
- Yamashita, A.; Sekizuka, T.; Kuroda, M. VirusTAP. Viral Genome-Targeted Assembly Pipeline. Front. Microbiol. 2016, 7, 32. [Google Scholar] [CrossRef]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Lurchachaiwong, W.; Junyangdikul, P.; Payungporn, S.; Chansaenroj, J.; Sampathanukul, P.; Tresukosol, D.; Termrungruanglert, W.; Theamboonlers, A.; Poovorawan, Y. Entire genome characterization of human papillomavirus type 16 from infected Thai women with different cytological findings. Virus Genes 2009, 39, 30–38. [Google Scholar] [CrossRef]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 2009, 537, 39–64. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Narisawa-Saito, M.; Inagawa, Y.; Yoshimatsu, Y.; Haga, K.; Tanaka, K.; Egawa, N.; Ohno, S.; Ichikawa, H.; Yugawa, T.; Fujita, M.; et al. A critical role of MYC for transformation of human cells by HPV16 E6E7 and oncogenic HRAS. Carcinogenesis 2012, 33, 910–917. [Google Scholar] [CrossRef]
- van der Weele, P.; Meijer, C.; King, A.J. Whole-genome sequencing and variant analysis of human papillomavirus 16 infections. J. Virol. 2017, 91, e00844-17. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; DeSalle, R.; Schiffman, M.; Herrero, R.; Wood, C.E.; Ruiz, J.C.; Clifford, G.M.; Chan, P.K.S.; Burk, R.D. Niche adaptation and viral transmission of human papillomaviruses from archaic hominins to modern humans. PLoS Pathog. 2018, 14, e1007352. [Google Scholar] [CrossRef] [PubMed]
- White, E.A.; Munger, K.; Howley, P.M. High-risk human papillomavirus E7 proteins target PTPN14 for degradation. mBio 2016, 7. [Google Scholar] [CrossRef]
- Szalmas, A.; Tomaic, V.; Basukala, O.; Massimi, P.; Mittal, S.; Konya, J.; Banks, L. The PTPN14 tumor suppressor is a degradation target of human papillomavirus E7. J. Virol. 2017, 91, e00057-17. [Google Scholar] [CrossRef]
- Matsumoto, K.; Yasugi, T.; Nakagawa, S.; Okubo, M.; Hirata, R.; Maeda, H.; Yoshikawa, H.; Taketani, Y. Human papillomavirus type 16 E6 variants and HLA class II alleles among Japanese women with cervical cancer. Int. J. Cancer 2003, 106, 919–922. [Google Scholar] [CrossRef]
- Ishizaki, A.; Matsushita, K.; Hoang, H.T.; Agdamag, D.M.; Nguyen, C.H.; Tran, V.T.; Sasagawa, T.; Saikawa, K.; Lihana, R.; Pham, H.V.; et al. E6 and E7 variants of human papillomavirus-16 and -52 in Japan, the Philippines, and Vietnam. J. Med. Virol. 2013, 85, 1069–1076. [Google Scholar] [CrossRef]
- Shang, Q.; Wang, Y.; Fang, Y.; Wei, L.; Chen, S.; Sun, Y.; Li, B.; Zhang, F.; Gu, H. Human papillomavirus type 16 variant analysis of E6, E7, and L1 genes and long control region in cervical carcinomas in patients in northeast China. J. Clin. Microbiol. 2011, 49, 2656–2663. [Google Scholar] [CrossRef]
- LeConte, B.A.; Szaniszlo, P.; Fennewald, S.M.; Lou, D.I.; Qiu, S.; Chen, N.W.; Lee, J.H.; Resto, V.A. Differences in the viral genome between HPV-positive cervical and oropharyngeal cancer. PLoS ONE 2018, 13, e0203403. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef]
- Aydin, I.; Villalonga-Planells, R.; Greune, L.; Bronnimann, M.P.; Calton, C.M.; Becker, M.; Lai, K.Y.; Campos, S.K.; Schmidt, M.A.; Schelhaas, M. A central region in the minor capsid protein of papillomaviruses facilitates viral genome tethering and membrane penetration for mitotic nuclear entry. PLoS Pathog. 2017, 13, e1006308. [Google Scholar] [CrossRef] [PubMed]
- Piersma, S.J.; Welters, M.J.; van der Hulst, J.M.; Kloth, J.N.; Kwappenberg, K.M.; Trimbos, B.J.; Melief, C.J.; Hellebrekers, B.W.; Fleuren, G.J.; Kenter, G.G.; et al. Human papilloma virus specific T cells infiltrating cervical cancer and draining lymph nodes show remarkably frequent use of HLA-DQ and -DP as a restriction element. Int. J. Cancer 2008, 122, 486–494. [Google Scholar] [CrossRef]
- Mirabello, L.; Yeager, M.; Yu, K.; Clifford, G.M.; Xiao, Y.; Zhu, B.; Cullen, M.; Boland, J.F.; Wentzensen, N.; Nelson, C.W.; et al. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 2017, 170, 1164–1174.e6. [Google Scholar] [CrossRef]
- Arroyo-Muhr, L.S.; Lagheden, C.; Hultin, E.; Eklund, C.; Adami, H.O.; Dillner, J.; Sundstrom, K. Human papillomavirus type 16 genomic variation in women with subsequent in situ or invasive cervical cancer: Prospective population-based study. Br. J. Cancer 2018, 119, 1163–1168. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Total | NILM | CIN1 | CIN2/3 | ICC |
|---|---|---|---|---|---|
| All | 100 | 22 | 13 | 24 | 41 |
| A | 94 | 21 | 11 | 24 | 38 |
| A1 | 21 | 3 | 3 | 12 | 3 |
| A2 | 2 | 0 | 2 | 0 | 0 |
| A3 | 3 | 1 | 0 | 0 | 2 |
| A4 | 52 | 10 | 4 | 8 | 30 |
| A5 | 16 | 7 | 2 | 4 | 3 |
| B | 0 | 0 | 0 | 0 | 0 |
| C | 2 | 0 | 1 | 0 | 1 |
| D | 4 | 1 | 1 | 0 | 2 |
| D1 | 1 | 0 | 1 | 0 | 0 |
| D2 | 1 | 0 | 0 | 0 | 1 |
| D3 | 2 | 1 | 0 | 0 | 1 |
| Variant | NILM/CIN1 | CIN2/3 | SCC | OR * (95% CI) | OR ** (95% CI) |
|---|---|---|---|---|---|
| All | 22 | 22 | 32 | ||
| A1/2/3 | 8 | 11 | 5 | 1.00 (reference) | 1.00 (reference) |
| A4 | 8 | 7 | 24 | 0.66 (0.16–2.62) | 6.72 (1.78–28.9) |
| A5 | 4 | 4 | 2 | 0.77 (0.13–4.41) | 0.07 (0.0003–1.83) |
| D1 | 1 | 0 | 0 | ND | ND |
| D3 | 1 | 0 | 1 | ND | ND |
| Region | Size (bp) | SNPs | Variable Sites (%) * | Synonymous | Non-Synonymous |
|---|---|---|---|---|---|
| All | 7905 ** | 142 | 1.80 | 57 | 60 |
| E6 | 477 | 6 | 1.26 | 3 | 3 |
| E7 | 297 | 4 | 1.35 | 4 | 0 |
| E1 | 1950 | 17 | 0.87 | 11 | 6 |
| E2 | 1098 | 17 | 1.55 | 4 | 13 |
| E4 | 288 | 7 | 2.43 | 4 | 3 |
| E5 | 252 | 9 | 3.57 | 1 | 8 |
| NC | 134 | 10 | 7.46 | ||
| L2 | 1422 | 37 | 2.60 | 16 | 21 |
| L1 | 1596 | 20 | 1.25 | 14 | 6 |
| LCR | 832 | 22 | 2.64 |
| Position * | A5 | Others | Gene | Position ** | A5 | Others |
|---|---|---|---|---|---|---|
| 645 | C | A | E7 | 28 | Phe | Leu |
| 3068 | A | G | E2 | 105 | Thr | Ala |
| 4458 | C | A | L2 | 74 | Pro | Pro |
| 5042 | A | C | L2 | 269 | Asp/Asn | Ser/Pro/Ala |
| 5836 | G | A | L1 | 92 | Ser | Ser |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirose, Y.; Onuki, M.; Tenjimbayashi, Y.; Yamaguchi-Naka, M.; Mori, S.; Tasaka, N.; Satoh, T.; Morisada, T.; Iwata, T.; Kiyono, T.; et al. Whole-Genome Analysis of Human Papillomavirus Type 16 Prevalent in Japanese Women with or without Cervical Lesions. Viruses 2019, 11, 350. https://doi.org/10.3390/v11040350
Hirose Y, Onuki M, Tenjimbayashi Y, Yamaguchi-Naka M, Mori S, Tasaka N, Satoh T, Morisada T, Iwata T, Kiyono T, et al. Whole-Genome Analysis of Human Papillomavirus Type 16 Prevalent in Japanese Women with or without Cervical Lesions. Viruses. 2019; 11(4):350. https://doi.org/10.3390/v11040350
Chicago/Turabian StyleHirose, Yusuke, Mamiko Onuki, Yuri Tenjimbayashi, Mayuko Yamaguchi-Naka, Seiichiro Mori, Nobutaka Tasaka, Toyomi Satoh, Tohru Morisada, Takashi Iwata, Tohru Kiyono, and et al. 2019. "Whole-Genome Analysis of Human Papillomavirus Type 16 Prevalent in Japanese Women with or without Cervical Lesions" Viruses 11, no. 4: 350. https://doi.org/10.3390/v11040350
APA StyleHirose, Y., Onuki, M., Tenjimbayashi, Y., Yamaguchi-Naka, M., Mori, S., Tasaka, N., Satoh, T., Morisada, T., Iwata, T., Kiyono, T., Mimura, T., Sekizawa, A., Matsumoto, K., & Kukimoto, I. (2019). Whole-Genome Analysis of Human Papillomavirus Type 16 Prevalent in Japanese Women with or without Cervical Lesions. Viruses, 11(4), 350. https://doi.org/10.3390/v11040350