A Reverse Genetics System for Cypovirus Based on a Bacmid Expressing T7 RNA Polymerase


Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses, Plasmids, Bacterial Strains, Cells, and Insects
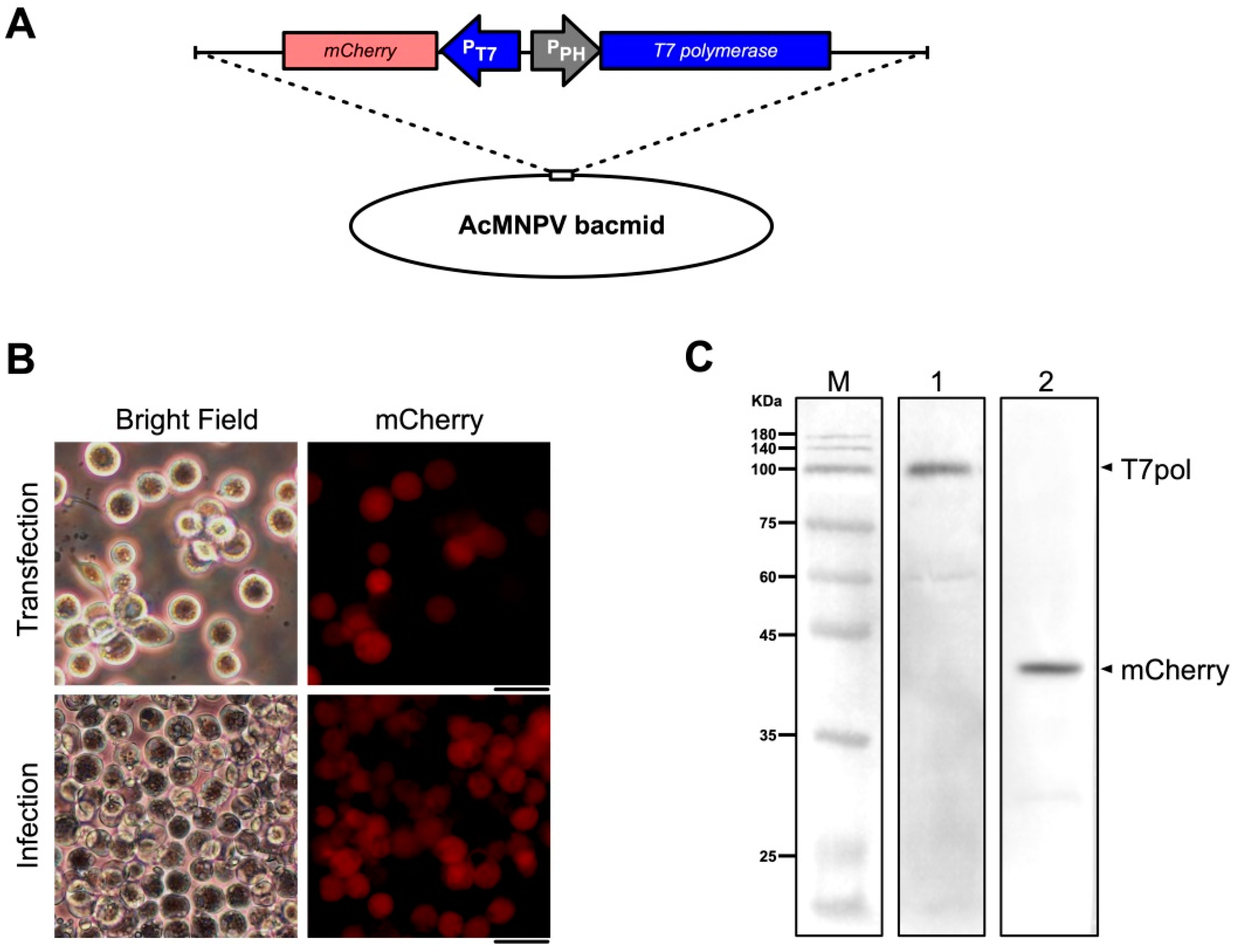
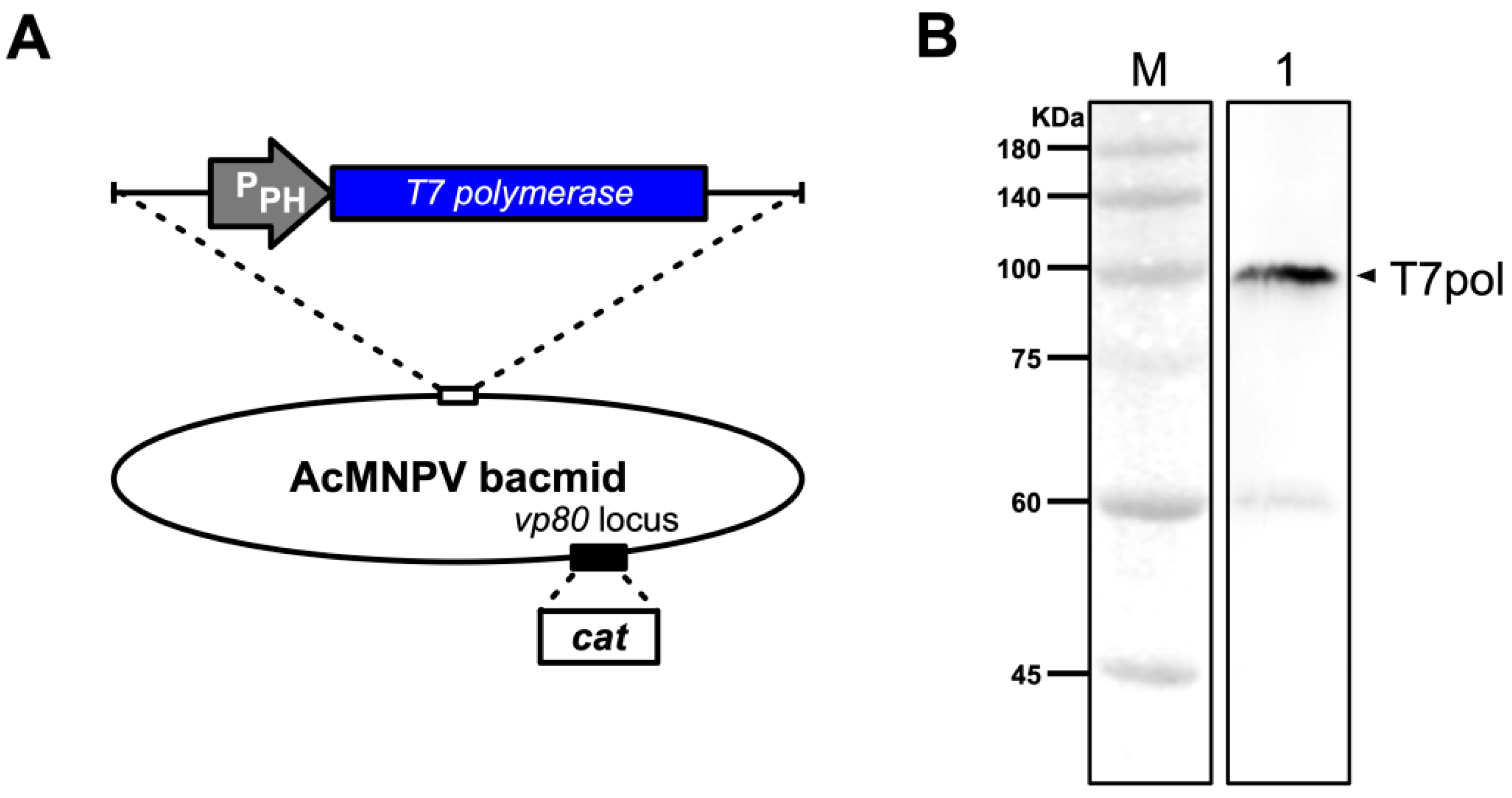
2.2. Construction and Identification of Recombinant Bacmids as A Source of T7 Polymerase
2.3. Construction of Three Recombinant Plasmids Bearing Full-Length cDNAs of DpCPV S1–S10 dsRNAs and An Additional Plasmid, T-S10UTR-egfp
2.4. Rescue of DpCPV OBs in Sf9 Cells
2.5. Identification of Rescued DpCPVs
2.6. Insecticidal Bioassays
2.7. Reverse Transcription-Quantitative PCR (RT-qPCR)
2.8. Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM)
2.9. Western Blotting
3. Results
3.1. A Codon-Optimized T7 RNA Polymerase Can be Expressed in Sf9 Cells
3.2. OBs Could be Recovered by Co-Transfection of T7 RNA Polymerase-Expressing Bacmid and Three Plasmids Containing cDNAs Derived from the 10 dsRNA Segments of DpCPV
3.3. Morphological Comparison and Detection of Genetic Markers Confirmed that DpCPVs were Correctly Rescued
3.4. Exogenous RNA Segments Might be Incorporated into CPV Particles
3.5. Infectivity of the Rescued DpCPVs was Lower than that of DpCPV-WT
3.6. Genetic Stability of the Rescued DpCPVs when Passaged in Host Insects
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- King, A.M.; Lefkowitz, E.J. Virus taxonomy. In IXth Report of the International Committee on Taxonomy of Viruses; Academic Press: New York, NY, USA, 2011; pp. 498–650. [Google Scholar]
- Chiu, E.; Coulibaly, F.; Metcalf, P. Insect virus polyhedra, infectious protein crystals that contain virus particles. Curr. Opin. Struct. Biol. 2012, 22, 234–240. [Google Scholar] [CrossRef]
- Xia, Q.; Jakana, J.; Zhang, J.Q.; Zhou, Z.H. Structural comparisons of empty and full cytoplasmic polyhedrosis virus. Protein-RNA interactions and implications for endogenous RNA transcription mechanism. J. Biol. Chem. 2003, 278, 1094–1100. [Google Scholar] [CrossRef]
- Nibert, M.L.; Baker, T.S. CPV, a Stable and Symmetrical Machine for mRNA Synthesis. Structure 2003, 11, 605–607. [Google Scholar] [CrossRef]
- Harrison, R.; Hoover, K. Baculoviruses and Other Occluded Insect Viruses. Insect Pathol. 2012, 1, 73–131. [Google Scholar]
- Belloncik, S. Interactions of Cytoplasmic Polyhedrosis Viruses with Insects. Adv. Insect Physiol. 1996, 26, 233–296. [Google Scholar]
- Kunimi, Y. Current status and prospects on microbial control in Japan. J. Invertebr. Pathol. 2007, 95, 181–186. [Google Scholar] [CrossRef]
- Sun, X. History and current status of development and use of viral insecticides in China. Viruses 2015, 7, 306–319. [Google Scholar] [CrossRef]
- Hagiwara, K.; Rao, S.; Scott, S.W.; Carmer, J.R. Nucleotide sequences of segments 1, 3 and 4 of the genome of Bombyx mori cypovirus 1 encoding putative capsid proteins VP1, VP3 and VP4, respectively. J. Gen. Virol. 2003, 148, 1357–1360. [Google Scholar] [CrossRef]
- Jin, L.; Dai, C.; Qin, T.; Sun, X. Molecular characterization of protein p50 of Dendrolimus punctatus cytoplasmic polyhedrosis virus. J. Basic Microbiol. 2013, 53, 37–44. [Google Scholar] [CrossRef]
- Rao, S.; Carner, G.R.; Scott, S.W.; Omura, T.; Hagiwara, K. Comparison of the amino acid sequences of RNA-dependent RNA polymerases of cypoviruses in the family Reoviridae. Arch. Virol. 2003, 148, 209–219. [Google Scholar] [CrossRef]
- Kaustubha, R.; Qanungo, S.C.K.; Mullins, J.; Ghosh, K. Molecular cloning and characterization of Antheraea mylitta cytoplasmic polyhedrosis virus genome segment 9. J. Gen. Virol. 2002, 83, 1483–1491. [Google Scholar]
- Sinha-Datta, U.; Chavali, V.R.; Ghosh, A.K. Molecular cloning and characterization of Antheraea mylitta cytoplasmic polyhedrosis virus polyhedrin gene and its variant forms. Biochem. Biophys. Res. Commun. 2005, 332, 710–718. [Google Scholar] [CrossRef]
- Liu, F.; Liu, Q.; Cai, Y.; Leng, Q.; Huang, Z. Construction and characterization of an infectious clone of coxsackievirus A16. Virol. J. 2011, 8, 534. [Google Scholar] [CrossRef]
- Takenaka-Uema, A.; Sugiura, K.; Bangphoomi, N.; Shioda, C.; Uchida, K.; Kato, K.; Haga, T.; Murakami, S.; Akashi, H.; Horimoto, T. Development of an improved reverse genetics system for Akabane bunyavirus. J. Virol. Methods 2016, 232, 16–20. [Google Scholar] [CrossRef]
- Almazán, F.; Sola, I.; Zuñiga, S.; Marquez-Jurado, S.; Morales, L.; Becares, M.; Enjuanes, L. Coronavirus reverse genetic systems: Infectious clones and replicons. Virus Res. 2014, 189, 262–270. [Google Scholar] [CrossRef]
- Li, P.; Ke, X.; Wang, T.; Tan, Z.; Luo, D.; Miao, Y.; Sun, J.; Zhang, Y.; Liu, Y.; Hu, Q. Zika Virus Attenuation by Codon Pair Deoptimization Induces Sterilizing Immunity in Mouse Models. J. Virol. 2018, 92, e00701-18. [Google Scholar] [CrossRef]
- Hu, B.; Jiang, J.; Zhan, J.; Li, G.; Jiang, Y.; Guan, X.; Chen, Y.; Fang, Z. Development of a reverse genetics system for respiratory syncytial virus long strain and an immunogenicity study of the recombinant virus. Virol. J. 2014, 11, 142. [Google Scholar] [CrossRef]
- Trask, S.D.; Taraporewala, Z.F.; Boehme, K.W.; Dermody, T.S.; Patton, J.T. Dual selection mechanisms drive efficient single-gene reverse genetics for rotavirus. Proc. Natl. Acad. Sci. USA 2010, 107, 18652–18657. [Google Scholar] [CrossRef]
- Kobayashi, T.; Antar, A.A.R.; Boehme, K.W.; Danthi, P.; Eby, E.A.; Guglielmi, K.M.; Holm, G.H.; Johnson, E.M.; Maginnis, M.S.; Naik, S.; et al. A Plasmid-Based Reverse Genetics System for Animal Double-Stranded RNA Viruses. Cell Host Microbe 2007, 1, 147–157. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ooms, L.S.; Ikizler, M.; Chappell, J.D.; Dermody, T.S. An improved reverse genetics system for mammalian orthoreoviruses. Virology 2010, 398, 194–200. [Google Scholar] [CrossRef]
- Kanai, Y.; Komoto, S.; Kawagishi, T.; Nouda, R.; Nagasawa, N.; Onishi, M.; Matsuura, Y.; Taniguchi, K.; Kobayashi, T. Entirely plasmid-based reverse genetics system for rotaviruses. Proc. Natl. Acad. Sci. USA 2017, 114, 2349–2354. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, J.M.; Huismans, H.; Theron, J. Establishment of an entirely plasmid-based reverse genetics system for Bluetongue virus. Virology 2015, 486, 71–77. [Google Scholar] [CrossRef]
- Komoto, S.; Fukuda, S.; Ide, T.; Ito, N.; Sugiyama, M.; Yoshikawa, T.; Murata, T.; Taniguchi, K.; López, S. Generation of Recombinant Rotaviruses Expressing Fluorescent Proteins by Using an Optimized Reverse Genetics System. J. Virol. 2018, 92, e00588-18. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Cao, G.; Xue, R.; Kumar, D.; Chen, F.; Liu, W.; Jiang, Y.; Lu, Y.; Zhu, L.; Liang, Z.; et al. Exogenous gene can be expressed by a recombinant Bombyx mori cypovirus. Appl. Microbiol. Biotechnol. 2017, 102, 1367–1379. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Xu, C.; Hu, J.; Lei, C.; Zheng, Z.; Peng, K.; Wang, H.; Sun, X. Dissecting the cell entry pathway of baculovirus by single particle tracking and quantitative electron microscopic analysis. J. Virol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wen, A.; Shen, B.; Lu, J.; Huang, Y.; Chang, Y. FastCloning: A highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotechnol. 2011, 11, 92. [Google Scholar] [CrossRef]
- Cheng, C.G.; S, Y.; Su, L.; Zhou, Y.; Sun, X. Interactions among Dendrolimus punctatus cypovirus proteins and identification of the genomic segment encoding its A-spike. J. Gen. Virol. 2014, 95, 1532–1538. [Google Scholar] [CrossRef]
- Zhou, Y.; Qin, T.; Xiao, Y.; Qin, F.; Lei, C.; Sun, X. Genomic and Biological Characterization of a New Cypovirus Isolated from Dendrolimus punctatus. PLoS ONE 2014, 9, e113201. [Google Scholar] [CrossRef]
- Li, J.; Zhou, Y.; Lei, C.; Fang, W.; Sun, X. Improvement in the UV resistance of baculoviruses by displaying nano-zinc oxide-binding peptides on the surfaces of their occlusion bodies. Appl. Microbiol. Biotechnol. 2015, 99, 6841–6853. [Google Scholar] [CrossRef]
- Norusis, M. SPSS 13.0 Advanced Statistical Procedures Companion; Prentice Hall, Inc.: Upper Saddle-River, NJ, USA, 2004. [Google Scholar]
- Robertson, J.L.; Jones, M.M.; Olguin, E.; Alberts, B. Bioassays with Arthropods, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Taylor, S.; Wakem, M.; Dijkman, G.; Alsarraj, M.; Nguyen, M. A practical approach to RT-qPCR—Publishing data that conform to the MIQE guidelines. Methods 2010, 50, S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Robert, L.; Harrison, M.D.S. Mutations in the Autographa californica multinucleocapsid nuclear polyhedrosis virus 25 kDa protein gene result in reduced virion occlusion, altered intranuclear envelopment and enhanced virus production. J. Gen. Virol. 1995, 1451–1459. [Google Scholar]
- Marek, M.; van Oers, M.M.; Devaraj, F.F.; Vlak, J.M.; Merten, O.-W. Engineering of baculovirus vectors for the manufacture of virion-free biopharmaceuticals. Biotechnol. Bioeng. 2011, 108, 1056–1067. [Google Scholar] [CrossRef]
- Theilmann, D.A.; Stewart, S. Identification and Characterization of the IE-1 Gene of Orgyia pseudotsugata Multicapsid Nuclear Polyhedrosis Virus. Virology 1991, 180, 492–508. [Google Scholar] [CrossRef]
- Theilmann, D.A.; Stewart, S. Molecular Analysis of the trans-Activating IE-2 Gene of Orgyia pseudotsugata Multicapsid Nuclear Polyhedrosis Virus. Virology 1992, 187, 84–96. [Google Scholar] [CrossRef]
- Bunch, T.A.; Grinblat, Y.; Goldstein, L.S. Characterization and use of the Drosophila metallothionein promoter in cultured Drosophila melanogaster cells. Nucleic Acids Res. 1988, 16, 1043–1061. [Google Scholar] [CrossRef] [PubMed]
- Shirk, P.D.; Furlong, R.B. Insect cell transformation vectors that support high level expression and promoter assessment in insect cell culture. Plasmid 2016, 83, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-X.; Wei, T.; Li, X.-Y.; Yin, X.; Li, Y.-H.; Ding, J.-W.; Zhou, J.-M.; Zhang, G.-Z.; Jin, Q.; Cen, S. Construction and characterization of an infectious cDNA clone of enterovirus type 71 subgenotype C4. Virus Genes 2013, 47, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Trask, S.D.; Patton, J.T. Generation of genetically stable recombinant rotaviruses containing novel genome rearrangements and heterologous sequences by reverse genetics. J. Virol. 2013, 87, 6211–6220. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, J.; Yu, X.; Lu, X.; Zhang, Q.; Jakana, J.; Chen, D.H.; Zhang, X.; Zhou, Z.H. Visualization of Protein-RNA Interactions in Cytoplasmic Polyhedrosis Virus. J. Virol. 1999, 73, 1624–1629. [Google Scholar] [PubMed]
- Ciechonska, M.; Duncan, R. Reovirus FAST proteins: Virus-encoded cellular fusogens. Trends Microbiol. 2014, 22, 715–724. [Google Scholar] [CrossRef]
- Yang, S.; Zhao, L.; Ma, R.; Fang, W.; Hu, J.; Lei, C.; Sun, X. Improving baculovirus infectivity by efficiently embedding enhancing factors into occlusion bodies. Appl. Environ. Microbiol. 2017, 83, 00595-17. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′–3′) a,b |
|---|---|
| F-HindIII-T7pol | CCCAAGCTTATGAACACAATCAACATCGCTAAGAACG |
| R-BamHI-T7pol | CGCGGATCCTTAAGCGAAGGCGAAGTCGGACTC |
| F-pFD-delp10 | CGTATACTCCGGAATATTAATAGATC |
| R-pFD-delp10 | CGGCAATAAAAAGACAGAATAAAACG |
| F-T7pro-mCherry | TATTCCGGAGTATACGTAATACGACTCACTATAGGGCGCC |
| R-mCherry | TGTCTTTTTATTGCCGCTTGTACAGCTCGTCCATGCCG |
| F-T-T7pro | ATAGTGAGTCGTATTAATCTCTGGAAGATCCGCGCGTACC |
| R-T-HDV | GGGTCGGCATGGCATCTCCA |
| F-T7pro-S1 | TAATACGACTCACTATAGAGTAAAGTGTATGTCTATACCAGAA |
| F-T7pro-S2 | TAATACGACTCACTATAGAGTAAAAGTCAGTATCTTACCGGCA |
| F-T7pro-S3 | TAATACGACTCACTATAGAGTAAAGACAGATGACGAGAGAC |
| F-T7pro-S4 | TAATACGACTCACTATAGAGTAATTTCCACCATGTGGCATTATAC |
| F-T7pro-S5 | TAATACGACTCACTATAGAGTAATTTCCCCGTCACTTAAAGG |
| F-T7pro-S6 | TAATACGACTCACTATAGAGTAAGATTCCGCAATATCCCATGG |
| F-T7pro-S7 | TAATACGACTCACTATAGAGTAATTTGGTCATAACAGCAAAG |
| F-T7pro-S8 | TAATACGACTCACTATAGAGTAAAGTCCAGTACTAGTTAAAGAC |
| F-T7pro-S9 | TAATACGACTCACTATAGAGGAAATCCCAGGTGTAAACCGAAT |
| F-T7pro-S10 | TAATACGACTCACTATAGAGTAAAAGTCAGTATCTTACCGGCA |
| R-S1-HDV | GAGATGCCATGCCGACCCGGCTAACGGTCGTGTATGAATGAGG |
| R-S2-HDV | GAGATGCCATGCCGACCCGGCTAACTCTGAACAGCGTACATC |
| R-S3-HDV | GAGATGCCATGCCGACCCGGCTAACGGTCGACACATGTTCATGC |
| R-S4-HDV | GAGATGCCATGCCGACCCGGCTAACGTTTCCCACCCC |
| R-S5-HDV | GAGATGCCATGCCGACCCGGCTAACCATCTCCCCGTG |
| R-S6-HDV | GAGATGCCATGCCGACCCGGCTAACGTTGACTCCGCTT |
| R-S7-HDV | GAGATGCCATGCCGACCCGGCTAACGTTTGGTCACTCCG |
| R-S8-HDV | GAGATGCCATGCCGACCCGGCTAACGGTAGTCCAGCCTGTTG |
| R-S9-HDV | GAGATGCCATGCCGACCCGGCTAACTACCCAGTGCCCTAAGG |
| R-S10-HDV | GAGATGCCATGCCGACCCGGCTAACTGTCAGTCAGTACCGC |
| F-T7ter-pBS | ATATCCGGATGGTACCGTCATAGCTGTTTCCTGTG |
| R-pBS-T7pro | ATAGTGAGTCGTATTACATGATTACGCCAAGCGC |
| R-T7ter | GGTACCATCCGGATATAGTTCCTCC |
| F-T7ter-pBS-2 | ATATCCGGATGGTACCTTGCGCGCTTGGCGTAATC |
| R-pBS-T7pro-2 | ATAGTGAGTCGTATTATTAACCCTCACTAAAGGGAACAAAAG |
| F-T7ter-pBS-3 | ATATCCGGATGGTACCGCTTTTGTTCCCTTTAGTGAGGG |
| R-pBS-T7pro-3 | ATAGTGAGTCGTATTATGGGTACCGGGCCCCCCC |
| F-T7ter-pBS-4 | ATATCCGGATGGTACCCCGCTCTAGAACTAGTGGATCCC |
| R-pBS-T7pro-4 | ATAGTGAGTCGTATTAGATCCACTAGTTCTAGAGCGGCCG |
| F-DpCPV-S9M | AGCTGGTAGAGTCTACTCCCCTGC |
| R-DpCPV-S9M | GTAGACTCTACCAGCTTCACGTCGG |
| F-egfp | TAATACGTAAAGGATCATGGTGAGCAAGGGCGAGG |
| R-egfp | CAAGTTACACGAGCAATTACTTGTACAGCTCGTCCATGCC |
| F-T-S10 | TTGCTCGTGTAACTTGGATACCAG |
| R-T-S10 | GATCCTTTACGTATTATGCCGG |
| Probe-qPCR-S2 c | GCTAGAAGTGGGAGGTGACGTAGCAGC |
| F-qPCR-S2 | TGAGGCATGGCTAAATTTCC |
| R-qPCR-S2 | AACCGCCTGCATAACAATTC |
| Probe-qPCR-S9 c | TTACGCCCAGCGCATCTCACCC |
| F-qPCR-S9 | TGGTATGGGTAAAATCAGGTCTTG |
| R-qPCR-S9 | TCGAGGATGCGAAATTTACATATG |
| Probe-qPCR-S10 c | ACTATCCTAATGGCGGCGACGCGCA |
| F-qPCR-S10 | CAAGGAGTATCGCGAAGGGC |
| R-qPCR-S10 | ATTTGGATCGCACGTGGCTT |
| Probe-qPCR-egfp c | AGGCTACGTCCAGGAGCGCACCATCTT |
| F-qPCR-egfp | CCACATGAAGCAGCACGACT |
| R-qPCR-egfp | GGGTCTTGTAGTTGCCGTCG |
| Virus | LC50 (95% CI) (×106 OBs/mL) | Potency Ratio (95% CI) to DpCPV-WT a |
|---|---|---|
| DpCPV-WT | 2.47 (0.97–8.06) | - |
| rDpCPV | 25.7 (7.6–75.3) | 11.4 (1.3–1586.5) |
| rDpCPV-S9M | >100 | |
| rDpCPV-egfp | >300 |
| Virus | Section Numbers | Solid Particles | Empty Particles | Ration | χ2 | p |
|---|---|---|---|---|---|---|
| DpCPV-WT | 7 | 268 | 205 | 1.307 | ||
| rDpCPV | 10 | 149 | 119 | 1.252 | 0.079 | 0.779 |
| rDpCPV-S9M | 14 | 206 | 167 | 1.233 | 0.174 | 0.677 |
| rDpCPV-egfp | 10 | 225 | 183 | 1.230 | 0.203 | 0.652 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, G.; Yang, J.; Qin, F.; Xu, C.; Wang, J.; Lei, C.; Hu, J.; Sun, X. A Reverse Genetics System for Cypovirus Based on a Bacmid Expressing T7 RNA Polymerase. Viruses 2019, 11, 314. https://doi.org/10.3390/v11040314
Zhang G, Yang J, Qin F, Xu C, Wang J, Lei C, Hu J, Sun X. A Reverse Genetics System for Cypovirus Based on a Bacmid Expressing T7 RNA Polymerase. Viruses. 2019; 11(4):314. https://doi.org/10.3390/v11040314
Chicago/Turabian StyleZhang, Gaobo, Jian Yang, Fujun Qin, Congrui Xu, Jia Wang, Chengfeng Lei, Jia Hu, and Xiulian Sun. 2019. "A Reverse Genetics System for Cypovirus Based on a Bacmid Expressing T7 RNA Polymerase" Viruses 11, no. 4: 314. https://doi.org/10.3390/v11040314
APA StyleZhang, G., Yang, J., Qin, F., Xu, C., Wang, J., Lei, C., Hu, J., & Sun, X. (2019). A Reverse Genetics System for Cypovirus Based on a Bacmid Expressing T7 RNA Polymerase. Viruses, 11(4), 314. https://doi.org/10.3390/v11040314