Antiviral Functions of Monoclonal Antibodies against Chikungunya Virus


Abstract
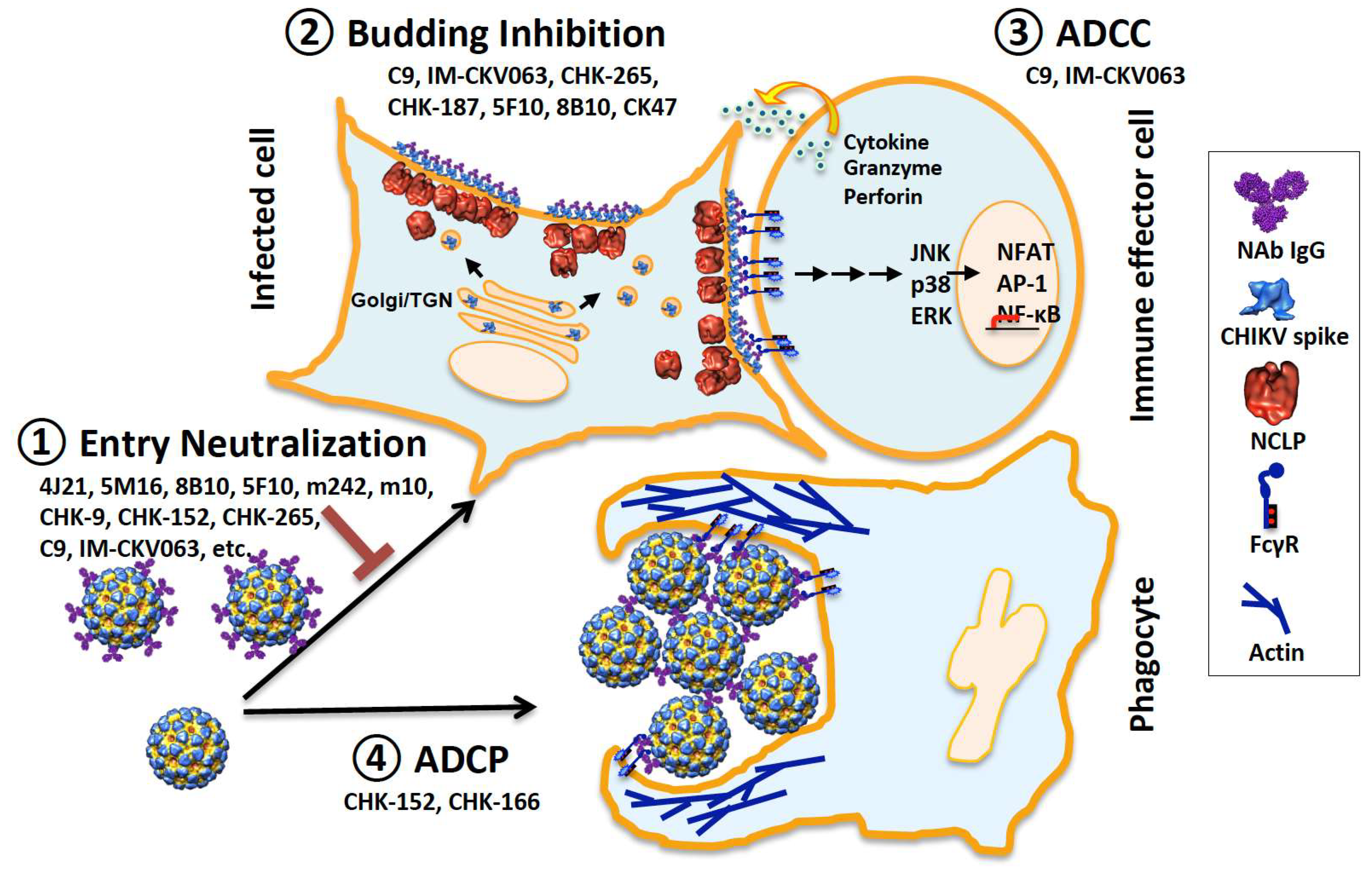
1. Introduction
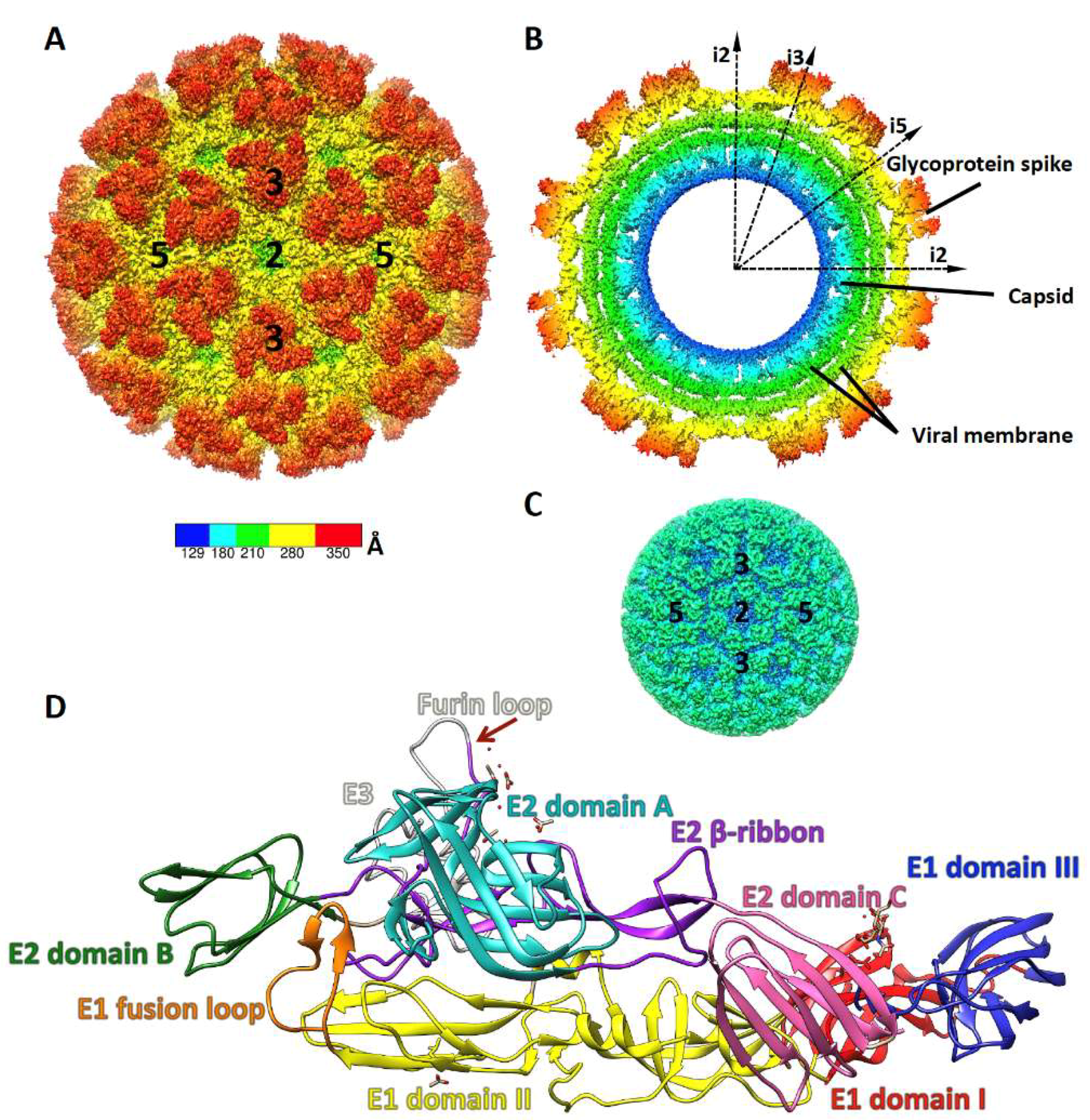
2. Alphavirus Structure and Life Cycle
3. Entry Neutralization by mAbs
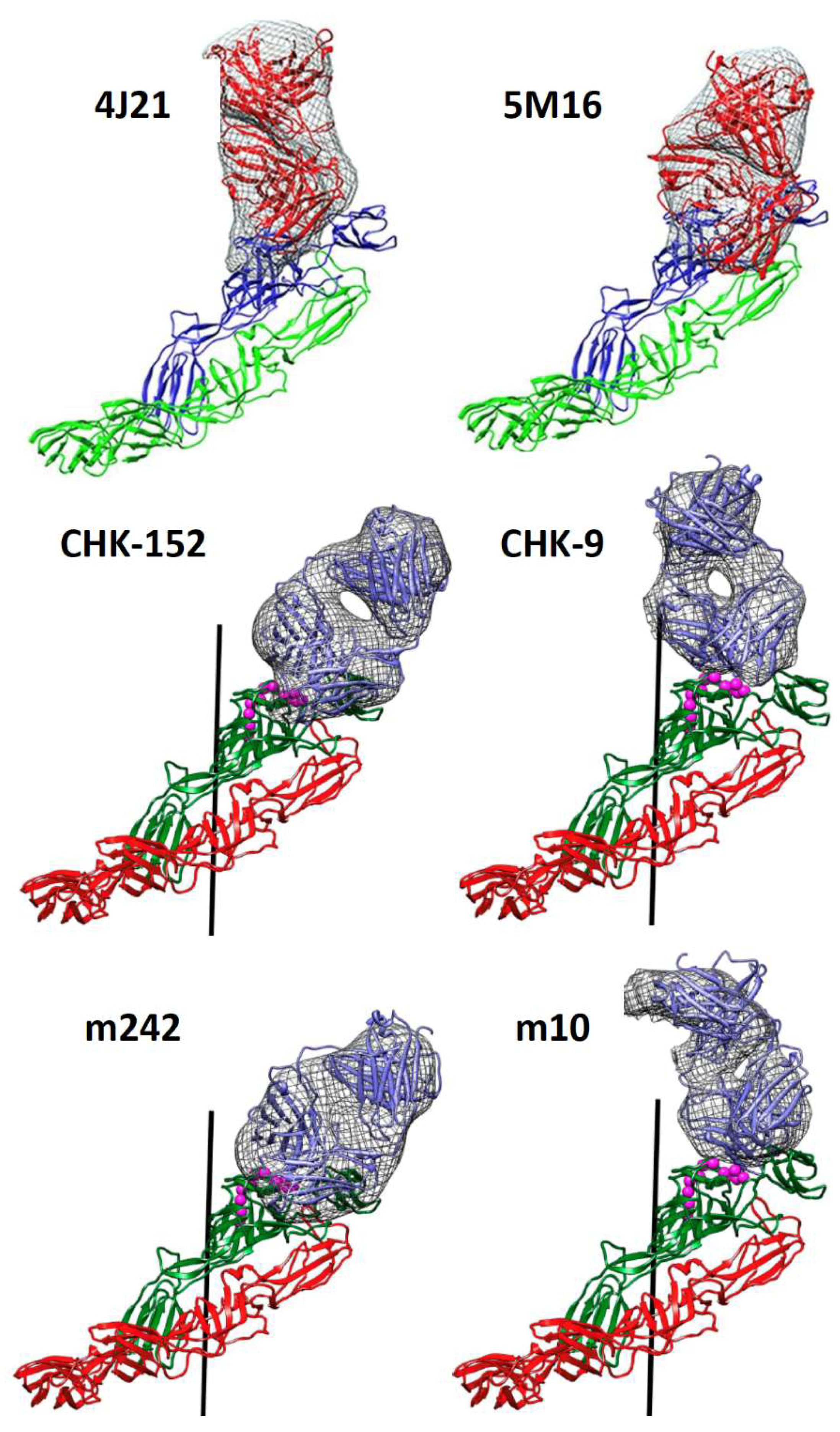
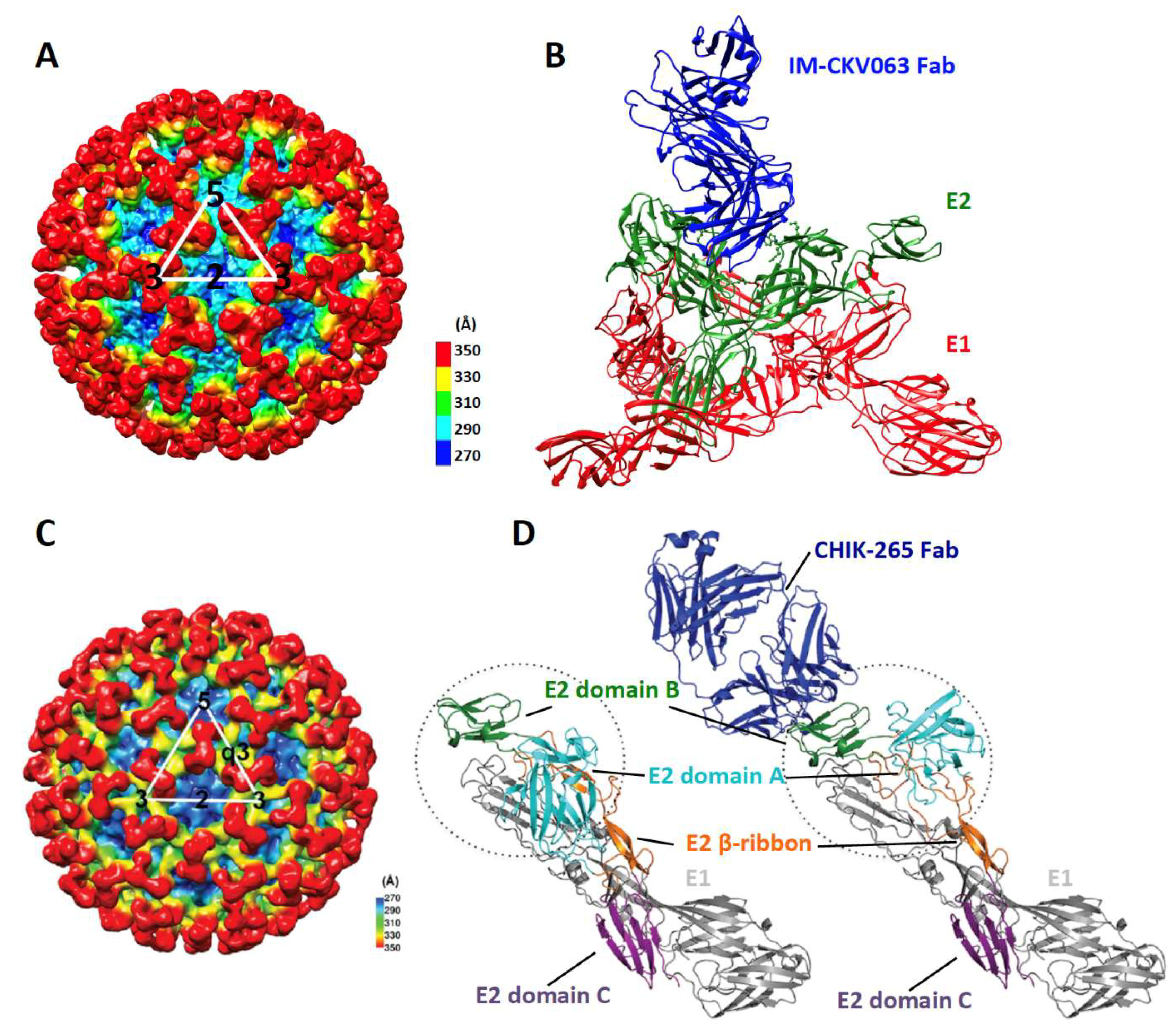
3.1. Block Virus Binding to Receptors
3.2. Prevent Activation of Membrane Fusion
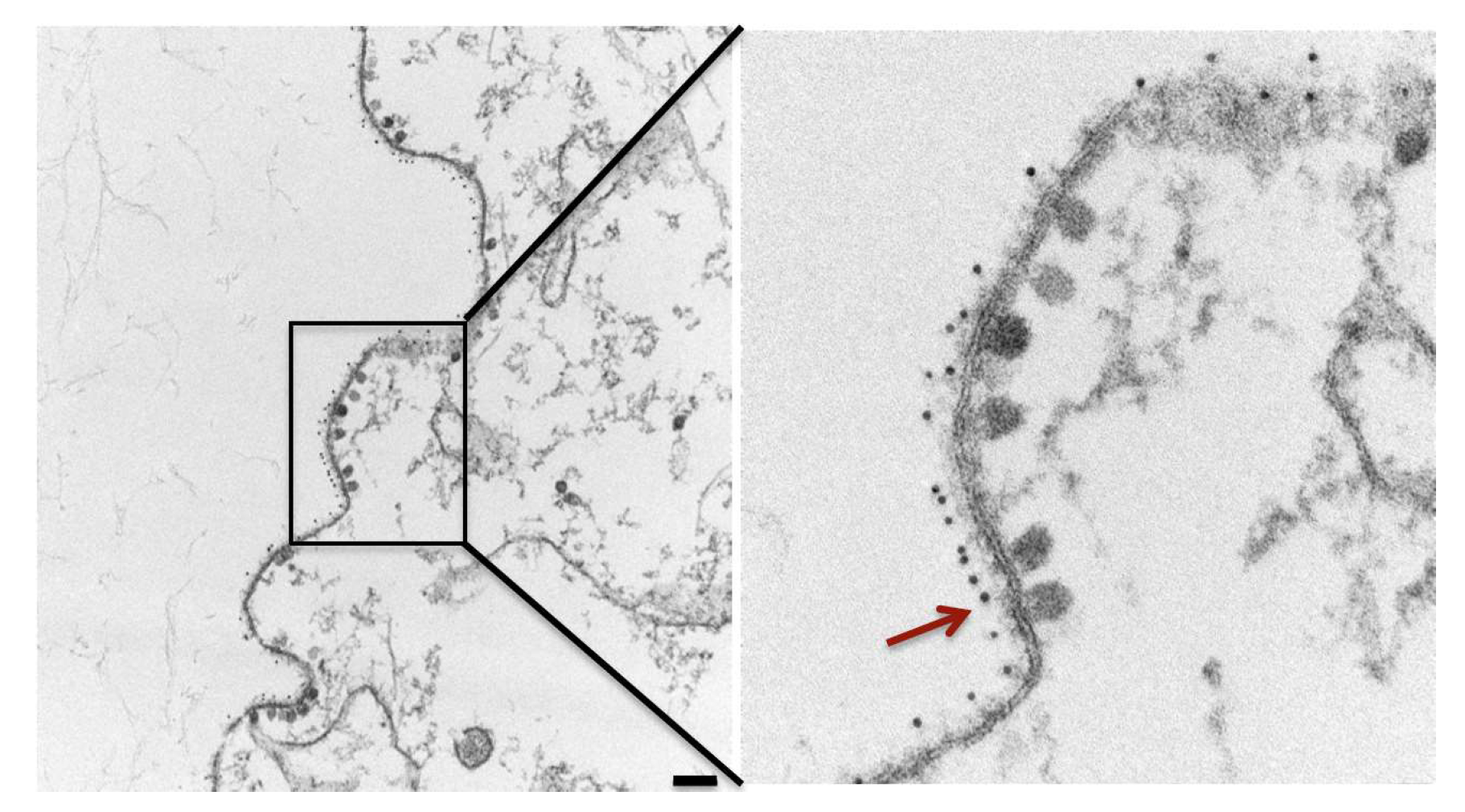
4. Budding Inhibition by mAbs
5. Antibody-Activated Effector Functions
6. Neutralizing IgM mAbs
7. Development of Antibody Therapeutics for CHIKV-Infection
8. Outstanding Questions and Conclusions
Funding
Conflicts of Interest
References
- Weaver, S.C.; Charlier, C.; Vasilakis, N.; Lecuit, M. Zika, chikungunya, and other emerging vector-borne viral diseases. Annu. Rev. Med. 2018, 69, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Couderc, T.; Lecuit, M. Chikungunya virus pathogenesis: From bedside to bench. Antivir. Res. 2015, 121, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Gopalan, S.S.; Das, A. Household economic impact of an emerging disease in terms of catastrophic out-of-pocket health care expenditure and loss of productivity: Investigation of an outbreak of chikungunya in orissa, india. J. Vector Borne Dis. 2009, 46, 57–64. [Google Scholar] [PubMed]
- Gerardin, P.; Couderc, T.; Bintner, M.; Tournebize, P.; Renouil, M.; Lemant, J.; Boisson, V.; Borgherini, G.; Staikowsky, F.; Schramm, F.; et al. Chikungunya virus-associated encephalitis: A cohort study on la reunion island, 2005–2009. Neurology 2015. [Google Scholar] [CrossRef] [PubMed]
- Fong, R.H.; Banik, S.S.; Mattia, K.; Barnes, T.; Tucker, D.; Liss, N.; Lu, K.; Selvarajah, S.; Srinivasan, S.; Mabila, M.; et al. Exposure of epitope residues on the outer face of the chikungunya virus envelope trimer determines antibody neutralizing efficacy. J. Virol. 2014, 88, 14364–14379. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.M.; Long, F.; Edeling, M.A.; Lin, H.; van Duijl-Richter, M.K.; Fong, R.H.; Kahle, K.M.; Smit, J.M.; Jin, J.; Simmons, G.; et al. Broadly neutralizing alphavirus antibodies bind an epitope on e2 and inhibit entry and egress. Cell 2015, 163, 1095–1107. [Google Scholar] [CrossRef]
- Jin, J.; Liss, N.M.; Chen, D.H.; Liao, M.; Fox, J.M.; Shimak, R.M.; Fong, R.H.; Chafets, D.; Bakkour, S.; Keating, S.; et al. Neutralizing monoclonal antibodies block chikungunya virus entry and release by targeting an epitope critical to viral pathogenesis. Cell Rep. 2015, 13, 2553–2564. [Google Scholar] [CrossRef]
- Pal, P.; Dowd, K.A.; Brien, J.D.; Edeling, M.A.; Gorlatov, S.; Johnson, S.; Lee, I.; Akahata, W.; Nabel, G.J.; Richter, M.K.; et al. Development of a highly protective combination monoclonal antibody therapy against chikungunya virus. PLoS Pathog. 2013, 9, e1003312. [Google Scholar] [CrossRef]
- Selvarajah, S.; Sexton, N.R.; Kahle, K.M.; Fong, R.H.; Mattia, K.A.; Gardner, J.; Lu, K.; Liss, N.M.; Salvador, B.; Tucker, D.F.; et al. A neutralizing monoclonal antibody targeting the acid-sensitive region in chikungunya virus e2 protects from disease. PLoS Negl. Trop. Dis. 2013, 7, e2423. [Google Scholar] [CrossRef]
- Smith, S.A.; Silva, L.A.; Fox, J.M.; Flyak, A.I.; Kose, N.; Sapparapu, G.; Khomadiak, S.; Ashbrook, A.W.; Kahle, K.M.; Fong, R.H.; et al. Isolation and characterization of broad and ultrapotent human monoclonal antibodies with therapeutic activity against chikungunya virus. Cell Host Microbe 2015, 18, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.; Nyo, M.; Phuektes, P.; Yew, C.W.; Tan, Y.J.; Chu, J.J. A potent neutralizing igm mab targeting the n218 epitope on e2 protein protects against chikungunya virus pathogenesis. MAbs 2015, 7, 1178–1194. [Google Scholar] [CrossRef] [PubMed]
- Warter, L.; Lee, C.Y.; Thiagarajan, R.; Grandadam, M.; Lebecque, S.; Lin, R.T.; Bertin-Maghit, S.; Ng, L.F.; Abastado, J.P.; Despres, P.; et al. Chikungunya virus envelope-specific human monoclonal antibodies with broad neutralization potency. J. Immunol. 2011, 186, 3258–3264. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Xiang, Y.; Akahata, W.; Holdaway, H.; Pal, P.; Zhang, X.; Diamond, M.S.; Nabel, G.J.; Rossmann, M.G. Structural analyses at pseudo atomic resolution of chikungunya virus and antibodies show mechanisms of neutralization. eLife 2013, 2, e00435. [Google Scholar] [CrossRef]
- Cheng, R.H.; Kuhn, R.J.; Olson, N.H.; Rossmann, M.G.; Choi, H.K.; Smith, T.J.; Baker, T.S. Nucleocapsid and glycoprotein organization in an enveloped virus. Cell 1995, 80, 621–630. [Google Scholar] [CrossRef]
- Zhang, R.; Hryc, C.F.; Cong, Y.; Liu, X.; Jakana, J.; Gorchakov, R.; Baker, M.L.; Weaver, S.C.; Chiu, W. 4.4 a cryo-em structure of an enveloped alphavirus venezuelan equine encephalitis virus. EMBO J. 2011, 30, 3854–3863. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, M.; Zhu, D.; Sun, Z.; Ma, J.; Wang, J.; Kong, L.; Wang, S.; Liu, Z.; Wei, L.; et al. Implication for alphavirus host-cell entry and assembly indicated by a 3.5a resolution cryo-em structure. Nat. Commun. 2018, 9, 5326. [Google Scholar] [CrossRef]
- Voss, J.E.; Vaney, M.C.; Duquerroy, S.; Vonrhein, C.; Girard-Blanc, C.; Crublet, E.; Thompson, A.; Bricogne, G.; Rey, F.A. Glycoprotein organization of chikungunya virus particles revealed by x-ray crystallography. Nature 2010, 468, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jose, J.; Xiang, Y.; Kuhn, R.J.; Rossmann, M.G. Structural changes of envelope proteins during alphavirus fusion. Nature 2010, 468, 705–708. [Google Scholar] [CrossRef]
- Dubuisson, J.; Rice, C.M. Sindbis virus attachment: Isolation and characterization of mutants with impaired binding to vertebrate cells. J. Virol. 1993, 67, 3363–3374. [Google Scholar]
- Zhang, R.; Kim, A.S.; Fox, J.M.; Nair, S.; Basore, K.; Klimstra, W.B.; Rimkunas, R.; Fong, R.H.; Lin, H.; Poddar, S.; et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 2018, 557, 570–574. [Google Scholar] [CrossRef]
- Lescar, J.; Roussel, A.; Wien, M.W.; Navaza, J.; Fuller, S.D.; Wengler, G.; Wengler, G.; Rey, F.A. The fusion glycoprotein shell of semliki forest virus: An icosahedral assembly primed for fusogenic activation at endosomal ph. Cell 2001, 105, 137–148. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Zhang, W.; Gabler, S.; Chipman, P.R.; Strauss, E.G.; Strauss, J.H.; Baker, T.S.; Kuhn, R.J.; Rossmann, M.G. Mapping the structure and function of the e1 and e2 glycoproteins in alphaviruses. Structure 2006, 14, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Vaney, M.C.; Roussel, A.; Vigouroux, A.; Reilly, B.; Lepault, J.; Kielian, M.; Rey, F.A. Conformational change and protein-protein interactions of the fusion protein of semliki forest virus. Nature 2004, 427, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Jose, J.; Snyder, J.E.; Kuhn, R.J. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009, 4, 837–856. [Google Scholar] [CrossRef]
- Choi, H.K.; Lu, G.; Lee, S.; Wengler, G.; Rossmann, M.G. Structure of semliki forest virus core protein. Proteins 1997, 27, 345–359. [Google Scholar] [CrossRef]
- Choi, H.K.; Tong, L.; Minor, W.; Dumas, P.; Boege, U.; Rossmann, M.G.; Wengler, G. Structure of sindbis virus core protein reveals a chymotrypsin-like serine proteinase and the organization of the virion. Nature 1991, 354, 37–43. [Google Scholar] [CrossRef]
- Jose, J.; Przybyla, L.; Edwards, T.J.; Perera, R.; Burgner, J.W., 2nd; Kuhn, R.J. Interactions of the cytoplasmic domain of sindbis virus e2 with nucleocapsid cores promote alphavirus budding. J. Virol. 2012, 86, 2585–2599. [Google Scholar] [CrossRef]
- Lee, S.; Owen, K.E.; Choi, H.K.; Lee, H.; Lu, G.; Wengler, G.; Brown, D.T.; Rossmann, M.G.; Kuhn, R.J. Identification of a protein binding site on the surface of the alphavirus nucleocapsid and its implication in virus assembly. Structure 1996, 4, 531–541. [Google Scholar] [CrossRef]
- Lamb, K.; Lokesh, G.L.; Sherman, M.; Watowich, S. Structure of a venezuelan equine encephalitis virus assembly intermediate isolated from infected cells. Virology 2010, 406, 261–269. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Chipman, P.R.; Hong, E.M.; Kuhn, R.J.; Rossmann, M.G. In vitro-assembled alphavirus core-like particles maintain a structure similar to that of nucleocapsid cores in mature virus. J. Virol. 2002, 76, 11128–11132. [Google Scholar] [CrossRef]
- Paredes, A.; Alwell-Warda, K.; Weaver, S.C.; Chiu, W.; Watowich, S.J. Structure of isolated nucleocapsids from venezuelan equine encephalitis virus and implications for assembly and disassembly of enveloped virus. J. Virol. 2003, 77, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Forsell, K.; Xing, L.; Kozlovska, T.; Cheng, R.H.; Garoff, H. Membrane proteins organize a symmetrical virus. EMBO J. 2000, 19, 5081–5091. [Google Scholar] [CrossRef] [PubMed]
- Forsell, K.; Griffiths, G.; Garoff, H. Preformed cytoplasmic nucleocapsids are not necessary for alphavirus budding. EMBO J. 1996, 15, 6495–6505. [Google Scholar] [CrossRef]
- Soonsawad, P.; Xing, L.; Milla, E.; Espinoza, J.M.; Kawano, M.; Marko, M.; Hsieh, C.; Furukawa, H.; Kawasaki, M.; Weerachatyanukul, W.; et al. Structural evidence of glycoprotein assembly in cellular membrane compartments prior to alphavirus budding. J. Virol. 2010, 84, 11145–11151. [Google Scholar] [CrossRef]
- Byrd, E.A.; Kielian, M. An alphavirus e2 membrane-proximal domain promotes envelope protein lateral interactions and virus budding. mBio 2017, 8. [Google Scholar] [CrossRef]
- Gomez-Navarro, N.; Miller, E.A. Cop-coated vesicles. Curr. Biol. 2016, 26, R54–R57. [Google Scholar] [CrossRef]
- Long, F.; Fong, R.H.; Austin, S.K.; Chen, Z.; Klose, T.; Fokine, A.; Liu, Y.; Porta, J.; Sapparapu, G.; Akahata, W.; et al. Cryo-em structures elucidate neutralizing mechanisms of anti-chikungunya human monoclonal antibodies with therapeutic activity. Proc. Natl Acad. Sci. USA 2015, 112, 13898–13903. [Google Scholar] [CrossRef]
- Fox, J.M.; Roy, V.; Gunn, B.M.; Huang, L.; Edeling, M.A.; Mack, M.; Fremont, D.H.; Doranz, B.J.; Johnson, S.; Alter, G.; et al. Optimal therapeutic activity of monoclonal antibodies against chikungunya virus requires fc-fc gamma recepot interaction on monocytes. Sci. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Kam, Y.W.; Fric, J.; Malleret, B.; Koh, E.G.; Prakash, C.; Huang, W.; Lee, W.W.; Lin, C.; Lin, R.T.; et al. Chikungunya virus neutralization antigens and direct cell-to-cell transmission are revealed by human antibody-escape mutants. PLoS Pathog. 2011, 7, e1002390. [Google Scholar] [CrossRef]
- Porta, J.; Jose, J.; Roehrig, J.T.; Blair, C.D.; Kuhn, R.J.; Rossmann, M.G. Locking and blocking the viral landscape of an alphavirus with neutralizing antibodies. J. Virol. 2014, 88, 9616–9623. [Google Scholar] [CrossRef]
- Jin, J.; Galaz-Montoya, J.G.; Sherman, M.B.; Sun, S.Y.; Goldsmith, C.S.; O’Toole, E.T.; Ackerman, L.; Carlson, L.A.; Weaver, S.C.; Chiu, W.; et al. Neutralizing antibodies inhibit chikungunya virus budding at the plasma membrane. Cell Host Microbe 2018, 24, 417–428 e415. [Google Scholar] [CrossRef] [PubMed]
- Masrinoul, P.; Puiprom, O.; Tanaka, A.; Kuwahara, M.; Chaichana, P.; Ikuta, K.; Ramasoota, P.; Okabayashi, T. Monoclonal antibody targeting chikungunya virus envelope 1 protein inhibits virus release. Virology 2014, 464–465, 111–117. [Google Scholar] [CrossRef]
- Porta, J.; Mangala Prasad, V.; Wang, C.I.; Akahata, W.; Ng, L.F.; Rossmann, M.G. Structural studies of chikungunya virus-like particles complexed with human antibodies: Neutralization and cell-to-cell transmission. J. Virol. 2016, 90, 1169–1177. [Google Scholar] [CrossRef]
- Sok, D.; Burton, D.R. Recent progress in broadly neutralizing antibodies to hiv. Nat. Immunol. 2018, 19, 1179–1188. [Google Scholar] [CrossRef]
- Corti, D.; Cameroni, E.; Guarino, B.; Kallewaard, N.L.; Zhu, Q.; Lanzavecchia, A. Tackling influenza with broadly neutralizing antibodies. Curr. Opin. Virol. 2017, 24, 60–69. [Google Scholar] [CrossRef]
- Bailey, J.R.; Flyak, A.I.; Cohen, V.J.; Li, H.; Wasilewski, L.N.; Snider, A.E.; Wang, S.; Learn, G.H.; Kose, N.; Loerinc, L.; et al. Broadly neutralizing antibodies with few somatic mutations and hepatitis c virus clearance. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Flyak, A.I.; Kuzmina, N.; Murin, C.D.; Bryan, C.; Davidson, E.; Gilchuk, P.; Gulka, C.P.; Ilinykh, P.A.; Shen, X.; Huang, K.; et al. Broadly neutralizing antibodies from human survivors target a conserved site in the ebola virus glycoprotein hr2-mper region. Nat. Microbiol. 2018, 3, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Wongwiwat, W.; Supasa, S.; Zhang, X.; Dai, X.; Rouvinski, A.; Jumnainsong, A.; Edwards, C.; Quyen, N.T.H.; Duangchinda, T.; et al. A new class of highly potent, broadly neutralizing antibodies isolated from viremic patients infected with dengue virus. Nat. Immunol. 2015, 16, 170–177. [Google Scholar] [CrossRef]
- Rouvinski, A.; Guardado-Calvo, P.; Barba-Spaeth, G.; Duquerroy, S.; Vaney, M.C.; Kikuti, C.M.; Navarro Sanchez, M.E.; Dejnirattisai, W.; Wongwiwat, W.; Haouz, A.; et al. Recognition determinants of broadly neutralizing human antibodies against dengue viruses. Nature 2015, 520, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Goh, L.Y.; Hobson-Peters, J.; Prow, N.A.; Gardner, J.; Bielefeldt-Ohmann, H.; Pyke, A.T.; Suhrbier, A.; Hall, R.A. Neutralizing monoclonal antibodies to the e2 protein of chikungunya virus protects against disease in a mouse model. Clin. Immunol. 2013, 149, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Fibriansah, G.; Tan, J.L.; Smith, S.A.; de Alwis, R.; Ng, T.S.; Kostyuchenko, V.A.; Jadi, R.S.; Kukkaro, P.; de Silva, A.M.; Crowe, J.E.; et al. A highly potent human antibody neutralizes dengue virus serotype 3 by binding across three surface proteins. Nat. Commun. 2015, 6, 6341. [Google Scholar] [CrossRef] [PubMed]
- Kam, Y.W.; Lum, F.M.; Teo, T.H.; Lee, W.W.; Simarmata, D.; Harjanto, S.; Chua, C.L.; Chan, Y.F.; Wee, J.K.; Chow, A.; et al. Early neutralizing igg response to chikungunya virus in infected patients targets a dominant linear epitope on the e2 glycoprotein. EMBO Mol. Med. 2012, 4, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Buchner, S.M.; Schnierle, B.S. A small antigenic determinant of the chikungunya virus e2 protein is sufficient to induce neutralizing antibodies which are partially protective in mice. PLoS Negl. Trop. Dis. 2015, 9, e0003684. [Google Scholar] [CrossRef] [PubMed]
- Erasmus, J.H.; Auguste, A.J.; Kaelber, J.T.; Luo, H.; Rossi, S.L.; Fenton, K.; Leal, G.; Kim, D.Y.; Chiu, W.; Wang, T.; et al. A chikungunya fever vaccine utilizing an insect-specific virus platform. Nat. Med. 2017, 23, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Partidos, C.D.; Weger, J.; Brewoo, J.; Seymour, R.; Borland, E.M.; Ledermann, J.P.; Powers, A.M.; Weaver, S.C.; Stinchcomb, D.T.; Osorio, J.E. Probing the attenuation and protective efficacy of a candidate chikungunya virus vaccine in mice with compromised interferon (ifn) signaling. Vaccine 2011, 29, 3067–3073. [Google Scholar] [CrossRef]
- Levitt, N.H.; Ramsburg, H.H.; Hasty, S.E.; Repik, P.M.; Cole, F.E., Jr.; Lupton, H.W. Development of an attenuated strain of chikungunya virus for use in vaccine production. Vaccine 1986, 4, 157–162. [Google Scholar] [CrossRef]
- Plante, K.; Wang, E.; Partidos, C.D.; Weger, J.; Gorchakov, R.; Tsetsarkin, K.; Borland, E.M.; Powers, A.M.; Seymour, R.; Stinchcomb, D.T.; et al. Novel chikungunya vaccine candidate with an ires-based attenuation and host range alteration mechanism. PLoS Pathog. 2011, 7, e1002142. [Google Scholar] [CrossRef] [PubMed]
- Akahata, W.; Yang, Z.Y.; Andersen, H.; Sun, S.; Holdaway, H.A.; Kong, W.P.; Lewis, M.G.; Higgs, S.; Rossmann, M.G.; Rao, S.; et al. A virus-like particle vaccine for epidemic chikungunya virus protects nonhuman primates against infection. Nat. Med. 2010, 16, 334–338. [Google Scholar] [CrossRef]
- Parker, M.D.; Buckley, M.J.; Melanson, V.R.; Glass, P.J.; Norwood, D.; Hart, M.K. Antibody to the e3 glycoprotein protects mice against lethal venezuelan equine encephalitis virus infection. J. Virol. 2010, 84, 12683–12690. [Google Scholar] [CrossRef]
- Dowdle, W.R.; Downie, J.C.; Laver, W.G. Inhibition of virus release by antibodies to surface antigens of influenza viruses. J. Virol. 1974, 13, 269–275. [Google Scholar]
- Yamayoshi, S.; Uraki, R.; Ito, M.; Kiso, M.; Nakatsu, S.; Yasuhara, A.; Oishi, K.; Sasaki, T.; Ikuta, K.; Kawaoka, Y. A broadly reactive human anti-hemagglutinin stem monoclonal antibody that inhibits influenza a virus particle release. EBio Med. 2017, 17, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, D.M.; Onuma, M.; Olson, C. Inhibition of bovine leukemia virus release by antiviral antibodies. Arch. Virol. 1977, 55, 139–144. [Google Scholar] [CrossRef]
- Shariff, D.M.; Davies, J.; Desperbasques, M.; Billstrom, M.; Geerligs, H.J.; Welling, G.W.; Welling-Wester, S.; Buchan, A.; Skinner, G.R. Immune inhibition of virus release from human and nonhuman cells by antibody to viral and host cell determinants. Intervirology 1991, 32, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Vanderplasschen, A.; Hollinshead, M.; Smith, G.L. Antibodies against vaccinia virus do not neutralize extracellular enveloped virus but prevent virus release from infected cells and comet formation. J. Gen.Virol. 1997, 78, 2041–2048. [Google Scholar] [CrossRef]
- Corboba, P.; Grutadauria, S.; Cuffini, C.; Zapata, M. Neutralizing monoclonal antibody to the e1 glycoprotein epitope of rubella virus mediates virus arrest in vero cells. Viral Immunol. 2000, 13, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Kajihara, M.; Marzi, A.; Nakayama, E.; Noda, T.; Kuroda, M.; Manzoor, R.; Matsuno, K.; Feldmann, H.; Yoshida, R.; Kawaoka, Y.; et al. Inhibition of marburg virus budding by nonneutralizing antibodies to the envelope glycoprotein. J. Virol. 2012, 86, 13467–13474. [Google Scholar] [CrossRef]
- Bournazos, S.; Ravetch, J.V. Fcgamma receptor pathways during active and passive immunization. Immunol. Rev. 2015, 268, 88–103. [Google Scholar] [CrossRef]
- Bournazos, S.; DiLillo, D.J.; Ravetch, J.V. The role of fc-fcgammar interactions in igg-mediated microbial neutralization. J. Exp. Med. 2015, 212, 1361–1369. [Google Scholar] [CrossRef]
- Bournazos, S.; Klein, F.; Pietzsch, J.; Seaman, M.S.; Nussenzweig, M.C.; Ravetch, J.V. Broadly neutralizing anti-hiv-1 antibodies require fc effector functions for in vivo activity. Cell 2014, 158, 1243–1253. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Palese, P.; Wilson, P.C.; Ravetch, J.V. Broadly neutralizing anti-influenza antibodies require fc receptor engagement for in vivo protection. J. Clin. Investig. 2016, 126, 605–610. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Tan, G.S.; Palese, P.; Ravetch, J.V. Broadly neutralizing hemagglutinin stalk-specific antibodies require fcgammar interactions for protection against influenza virus in vivo. Nat. Med. 2014, 20, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Gunn, B.M.; Yu, W.H.; Karim, M.M.; Brannan, J.M.; Herbert, A.S.; Wec, A.Z.; Halfmann, P.J.; Fusco, M.L.; Schendel, S.L.; Gangavarapu, K.; et al. A role for fc function in therapeutic monoclonal antibody-mediated protection against ebola virus. Cell Host Microbe 2018, 24, 221–233 e225. [Google Scholar] [CrossRef] [PubMed]
- Bruhns, P. Properties of mouse and human igg receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Analyzing antibody-fc-receptor interactions. Methods Mol. Biol. 2008, 415, 151–162. [Google Scholar]
- Lu, J.; Chu, J.; Zou, Z.; Hamacher, N.B.; Rixon, M.W.; Sun, P.D. Structure of fcgammari in complex with fc reveals the importance of glycan recognition for high-affinity igg binding. Proc. Natl. Acad. Sci. USA 2015, 112, 833–838. [Google Scholar] [CrossRef]
- Sondermann, P.; Huber, R.; Oosthuizen, V.; Jacob, U. The 3.2-a crystal structure of the human igg1 fc fragment-fc gammariii complex. Nature 2000, 406, 267–273. [Google Scholar] [CrossRef]
- Radaev, S.; Motyka, S.; Fridman, W.H.; Sautes-Fridman, C.; Sun, P.D. The structure of a human type iii fcgamma receptor in complex with fc. J. Biol. Chem. 2001, 276, 16469–16477. [Google Scholar] [CrossRef]
- Getahun, A.; Cambier, J.C. Of itims, itams, and itamis: Revisiting immunoglobulin fc receptor signaling. Immunol. Rev. 2015, 268, 66–73. [Google Scholar] [CrossRef]
- Goodridge, H.S.; Underhill, D.M.; Touret, N. Mechanisms of fc receptor and dectin-1 activation for phagocytosis. Traffic 2012, 13, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Sondermann, P.; Kaiser, J.; Jacob, U. Molecular basis for immune complex recognition: A comparison of fc-receptor structures. J. Mol. Biol. 2001, 309, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Jegaskanda, S.; Job, E.R.; Kramski, M.; Laurie, K.; Isitman, G.; de Rose, R.; Winnall, W.R.; Stratov, I.; Brooks, A.G.; Reading, P.C.; et al. Cross-reactive influenza-specific antibody-dependent cellular cytotoxicity antibodies in the absence of neutralizing antibodies. J. Immunol. 2013, 190, 1837–1848. [Google Scholar] [CrossRef] [PubMed]
- Jegaskanda, S.; Weinfurter, J.T.; Friedrich, T.C.; Kent, S.J. Antibody-dependent cellular cytotoxicity is associated with control of pandemic h1n1 influenza virus infection of macaques. J. Virol. 2013, 87, 5512–5522. [Google Scholar] [CrossRef] [PubMed]
- Saphire, E.O.; Schendel, S.L.; Fusco, M.L.; Gangavarapu, K.; Gunn, B.M.; Wec, A.Z.; Halfmann, P.J.; Brannan, J.M.; Herbert, A.S.; Qiu, X.; et al. Systematic analysis of monoclonal antibodies against ebola virus gp defines features that contribute to protection. Cell 2018, 174, 938–952 e913. [Google Scholar] [CrossRef] [PubMed]
- Seidel, U.J.; Schlegel, P.; Lang, P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front. Immunol. 2013, 4, 76. [Google Scholar] [CrossRef]
- Smith, P.; DiLillo, D.J.; Bournazos, S.; Li, F.; Ravetch, J.V. Mouse model recapitulating human fcgamma receptor structural and functional diversity. Proc. Natl. Acad. Sci. USA 2012, 109, 6181–6186. [Google Scholar] [CrossRef] [PubMed]
- DiLillo, D.J.; Ravetch, J.V. Differential fc-receptor engagement drives an anti-tumor vaccinal effect. Cell 2015, 161, 1035–1045. [Google Scholar] [CrossRef]
- Wang, X.; Mathieu, M.; Brezski, R.J. Igg fc engineering to modulate antibody effector functions. Protein Cell 2018, 9, 63–73. [Google Scholar] [CrossRef]
- Stavenhagen, J.B.; Gorlatov, S.; Tuaillon, N.; Rankin, C.T.; Li, H.; Burke, S.; Huang, L.; Johnson, S.; Koenig, S.; Bonvini, E. Enhancing the potency of therapeutic monoclonal antibodies via fc optimization. Adv. Enzyme Regul. 2008, 48, 152–164. [Google Scholar] [CrossRef]
- Jennewein, M.F.; Alter, G. The immunoregulatory roles of antibody glycosylation. Trends Immunol. 2017, 38, 358–372. [Google Scholar] [CrossRef]
- Diamond, M.S.; Sitati, E.M.; Friend, L.D.; Higgs, S.; Shrestha, B.; Engle, M. A critical role for induced igm in the protection against west nile virus infection. J. Exp. Med. 2003, 198, 1853–1862. [Google Scholar] [CrossRef]
- Dorfmeier, C.L.; Shen, S.; Tzvetkov, E.P.; McGettigan, J.P. Reinvestigating the role of igm in rabies virus postexposure vaccination. J. Virol. 2013, 87, 9217–9222. [Google Scholar] [CrossRef] [PubMed]
- Skountzou, I.; Satyabhama, L.; Stavropoulou, A.; Ashraf, Z.; Esser, E.S.; Vassilieva, E.; Koutsonanos, D.; Compans, R.; Jacob, J. Influenza virus-specific neutralizing igm antibodies persist for a lifetime. Clin. Vaccine Immunol. 2014, 21, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Moyron-Quiroz, J.E.; McCausland, M.M.; Kageyama, R.; Sette, A.; Crotty, S. The smallpox vaccine induces an early neutralizing igm response. Vaccine 2009, 28, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; Scallan, M.F.; Morris, M.M.; Dyson, H.; Fazakerley, J.K. Role of immune responses in protection and pathogenesis during semliki forest virus encephalitis. J. Gen. Virol. 1996, 77, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Nilaratanakul, V.; Chen, J.; Tran, O.; Baxter, V.K.; Troisi, E.M.; Yeh, J.X.; Griffin, D.E. Germ line igm is sufficient, but not required, for antibody-mediated alphavirus clearance from the central nervous system. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Pluschke, G.; Bordmann, G.; Daoudaki, M.E.; Lambris, J.D.; Achtman, M.; Neibert, M. Isolation of rat igm to igg hybridoma isotype switch variants and analysis of the efficiency of rat ig in complement activation. Eur. J. Immunol. 1989, 19, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Broeckel, R.; Fox, J.M.; Haese, N.; Kreklywich, C.N.; Sukulpovi-Petty, S.; Legasse, A.; Smith, P.P.; Denton, M.; Corvey, C.; Krishnan, S.; et al. Therapeutic administration of a recombinant human monoclonal antibody reduces the severity of chikungunya virus disease in rhesus macaques. PLoS Negl. Trop. Dis. 2017, 11, e0005637. [Google Scholar] [CrossRef]
- Pal, P.; Fox, J.M.; Hawman, D.W.; Huang, Y.J.; Messaoudi, I.; Kreklywich, C.; Denton, M.; Legasse, A.W.; Smith, P.P.; Johnson, S.; et al. Chikungunya viruses that escape monoclonal antibody therapy are clinically attenuated, stable, and not purified in mosquitoes. J. Virol. 2014, 88, 8213–8226. [Google Scholar] [CrossRef]
- Labadie, K.; Larcher, T.; Joubert, C.; Mannioui, A.; Delache, B.; Brochard, P.; Guigand, L.; Dubreil, L.; Lebon, P.; Verrier, B.; et al. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J. Clin. Investig. 2010, 120, 894–906. [Google Scholar] [CrossRef]
- Morrison, T.E.; Oko, L.; Montgomery, S.A.; Whitmore, A.C.; Lotstein, A.R.; Gunn, B.M.; Elmore, S.A.; Heise, M.T. A mouse model of chikungunya virus-induced musculoskeletal inflammatory disease: Evidence of arthritis, tenosynovitis, myositis, and persistence. Am. J. Pathol. 2011, 178, 32–40. [Google Scholar] [CrossRef]
- Appassakij, H.; Khuntikij, P.; Kemapunmanus, M.; Wutthanarungsan, R.; Silpapojakul, K. Viremic profiles in asymptomatic and symptomatic chikungunya fever: A blood transfusion threat? Transfusion 2013, 53, 2567–2574. [Google Scholar] [CrossRef] [PubMed]
- Kam, Y.W.; Simarmata, D.; Chow, A.; Her, Z.; Teng, T.S.; Ong, E.K.; Renia, L.; Leo, Y.S.; Ng, L.F. Early appearance of neutralizing immunoglobulin g3 antibodies is associated with chikungunya virus clearance and long-term clinical protection. J. Infect. Dis. 2012, 205, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.L.; Gilliland, G.L. Engineering antibody therapeutics. Curr. Opin. Struct. Biol. 2016, 38, 163–173. [Google Scholar] [CrossRef]
- Walker, L.M.; Burton, D.R. Passive immunotherapy of viral infections: ’Super-antibodies’ enter the fray. Nat. Rev. Immunol. 2018. [Google Scholar] [CrossRef]
- Klasse, P.J. Neutralization of virus infectivity by antibodies: Old problems in new perspectives. Adv. Biol. 2014. [Google Scholar] [CrossRef]
- Henry Dunand, C.J.; Leon, P.E.; Huang, M.; Choi, A.; Chromikova, V.; Ho, I.Y.; Tan, G.S.; Cruz, J.; Hirsh, A.; Zheng, N.Y.; et al. Both neutralizing and non-neutralizing human h7n9 influenza vaccine-induced monoclonal antibodies confer protection. Cell Host Microbe 2016, 19, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.K.; Finzi, A.; DeVico, A.L.; Pazgier, M. Conformational masking and receptor-dependent unmasking of highly conserved env epitopes recognized by non-neutralizing antibodies that mediate potent adcc against hiv-1. Viruses 2015, 7, 5115–5132. [Google Scholar] [CrossRef] [PubMed]
- Zeitlin, L.; Pettitt, J.; Scully, C.; Bohorova, N.; Kim, D.; Pauly, M.; Hiatt, A.; Ngo, L.; Steinkellner, H.; Whaley, K.J.; et al. Enhanced potency of a fucose-free monoclonal antibody being developed as an ebola virus immunoprotectant. Proc. Natl. Acad. Sci. USA 2011, 108, 20690–20694. [Google Scholar] [CrossRef]
- Qiu, X.; Audet, J.; Lv, M.; He, S.; Wong, G.; Wei, H.; Luo, L.; Fernando, L.; Kroeker, A.; Fausther Bovendo, H.; et al. Two-mab cocktail protects macaques against the makona variant of ebola virus. Sci. Transl. Med. 2016, 8, 329ra333. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mAb | Species | Antigenic Sites | CHIKV-LR EC50 ng/mL (CI) | Demonstrated Mode of Action | Reference |
|---|---|---|---|---|---|
| 2H1 | human | E2-DA | 5.6 (4.9–6.3) | entry neutralization | [10] |
| 8G18 | human | E2-DA | 7.3 (6.3–8.4) | entry neutralization | [10] |
| 3E23 | human | E2-DA | 18.7 (16.3–21.5) | entry neutralization | [10] |
| 1O6 | human | E2-DA | 57.5 (48.8–68.1) | entry neutralization | [10] |
| 4J21 | human | E2-DA, DB, arch | 7.4 (6.6–8.3) | entry neutralization | [10,37] |
| 5M16 | human | E2-DA, DB, arch | 5.9 (5.0–6.8) | entry neutralization | [10,37] |
| CHK-152 | mouse | E2-DA, DB, arch | 5 (5–7) | entry neutralization, ADCP | [8,12,13,38,39,40] |
| CHK-9 | mouse | E2-DA | 36 (31–43) | entry neutralization | [13,8] |
| CHK-166 | mouse | E1-DII | 154 (116–205) | entry neutralization, ADCP | [8,12,38,39,40] |
| CHK-265 | mouse | E2-DA, DB | 8 (7–9) | entry neutralization, viral egress inhibition | [6,8] |
| CHK-187 | mouse | E2-DB | 11 (9–12) | entry neutralization, viral egress inhibition | [6,8] |
| C9 | human | E2-DA, DB, arch | 51 * | entry neutralization, budding inhibition, ADCC | [7,9,41] |
| IM-CKV063 | human | E2-DA, DB, arch | 7.4 * | entry neutralization, budding inhibition, ADCC | [5,7,41] |
| m242 | human | E2-DA | 300 | entry neutralization | [13] |
| m10 | human | E2-DB | 7 | entry neutralization | [13] |
| 8B10 | human | E2-DA, DB | 46 # | entry neutralization, budding inhibition | [12,39,40] |
| 5F10 | human | E2-DB | 62 # | entry neutralization, budding inhibition | [12,39,40] |
| CK47 | mouse | E1-DIII | no | viral egress inhibition | [42] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, J.; Simmons, G. Antiviral Functions of Monoclonal Antibodies against Chikungunya Virus. Viruses 2019, 11, 305. https://doi.org/10.3390/v11040305
Jin J, Simmons G. Antiviral Functions of Monoclonal Antibodies against Chikungunya Virus. Viruses. 2019; 11(4):305. https://doi.org/10.3390/v11040305
Chicago/Turabian StyleJin, Jing, and Graham Simmons. 2019. "Antiviral Functions of Monoclonal Antibodies against Chikungunya Virus" Viruses 11, no. 4: 305. https://doi.org/10.3390/v11040305
APA StyleJin, J., & Simmons, G. (2019). Antiviral Functions of Monoclonal Antibodies against Chikungunya Virus. Viruses, 11(4), 305. https://doi.org/10.3390/v11040305