The CARD9-Associated C-Type Lectin, Mincle, Recognizes La Crosse Virus (LACV) but Plays a Limited Role in Early Antiviral Responses against LACV

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Virus and Cell Culture
2.3. Flow-Through Chromatography
2.4. CLR-hFc Fusion Protein Production
2.5. ELISA-Based LACV/CLR Binding Studies
2.6. Real-Time PCR
2.7. LACV Infection of BMDCs
2.8. Cytokine ELISAs
2.9. In Vitro Viral Replication
2.10. Transmission Electron Microscopy
2.11. Protein Concentration Determination
2.12. Statistical Analysis
3. Results
3.1. LACV Purification by Flow-through Chromatography
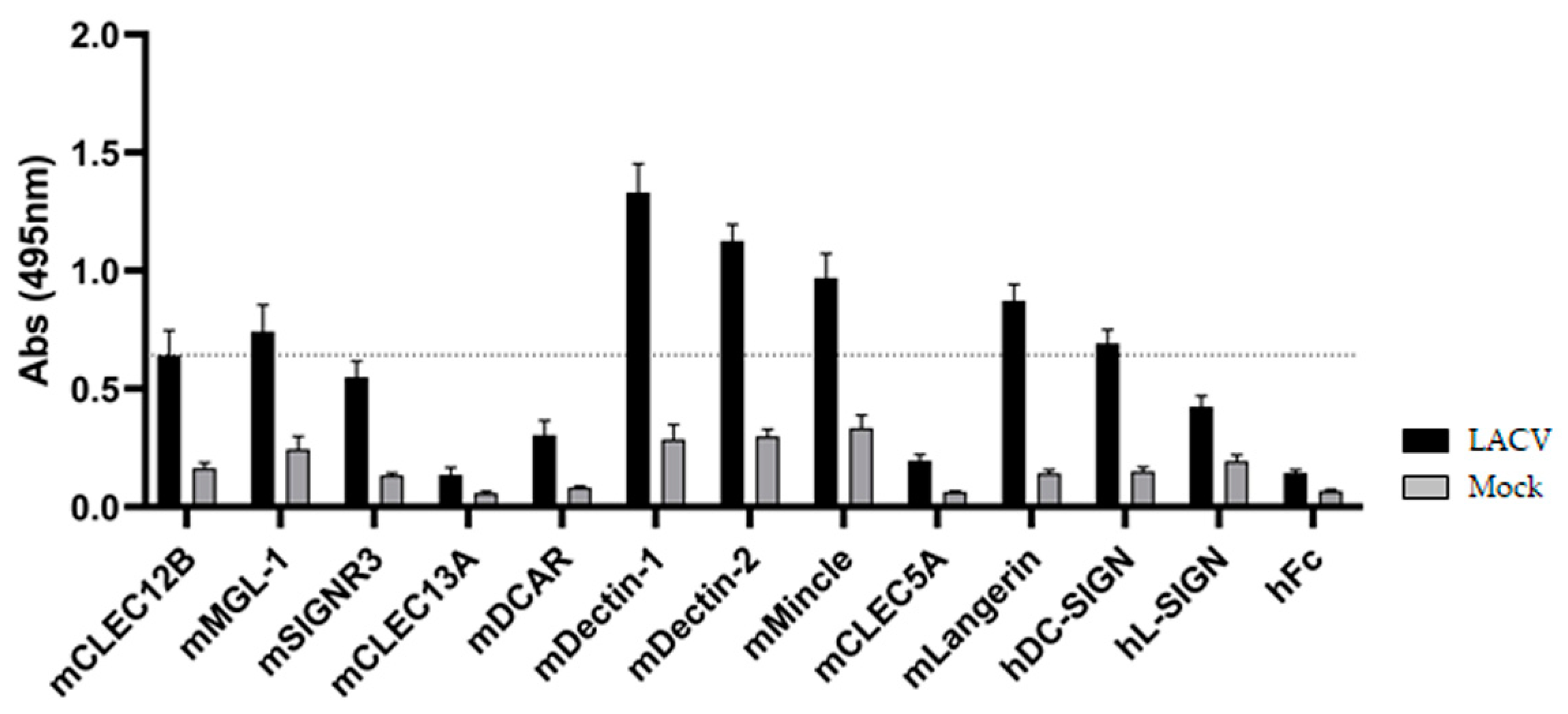
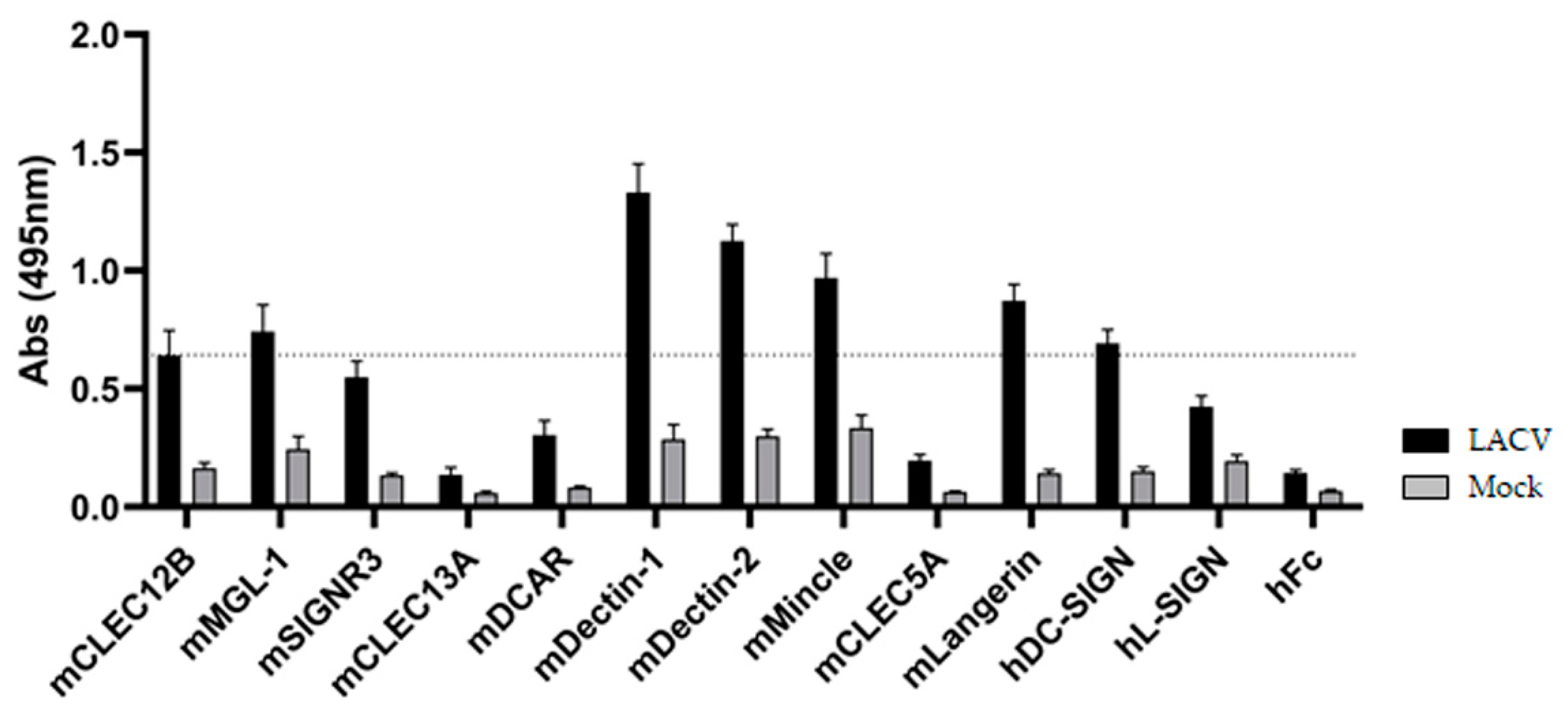
3.2. ELISA-Based Screening of LACV/CLR Interactions
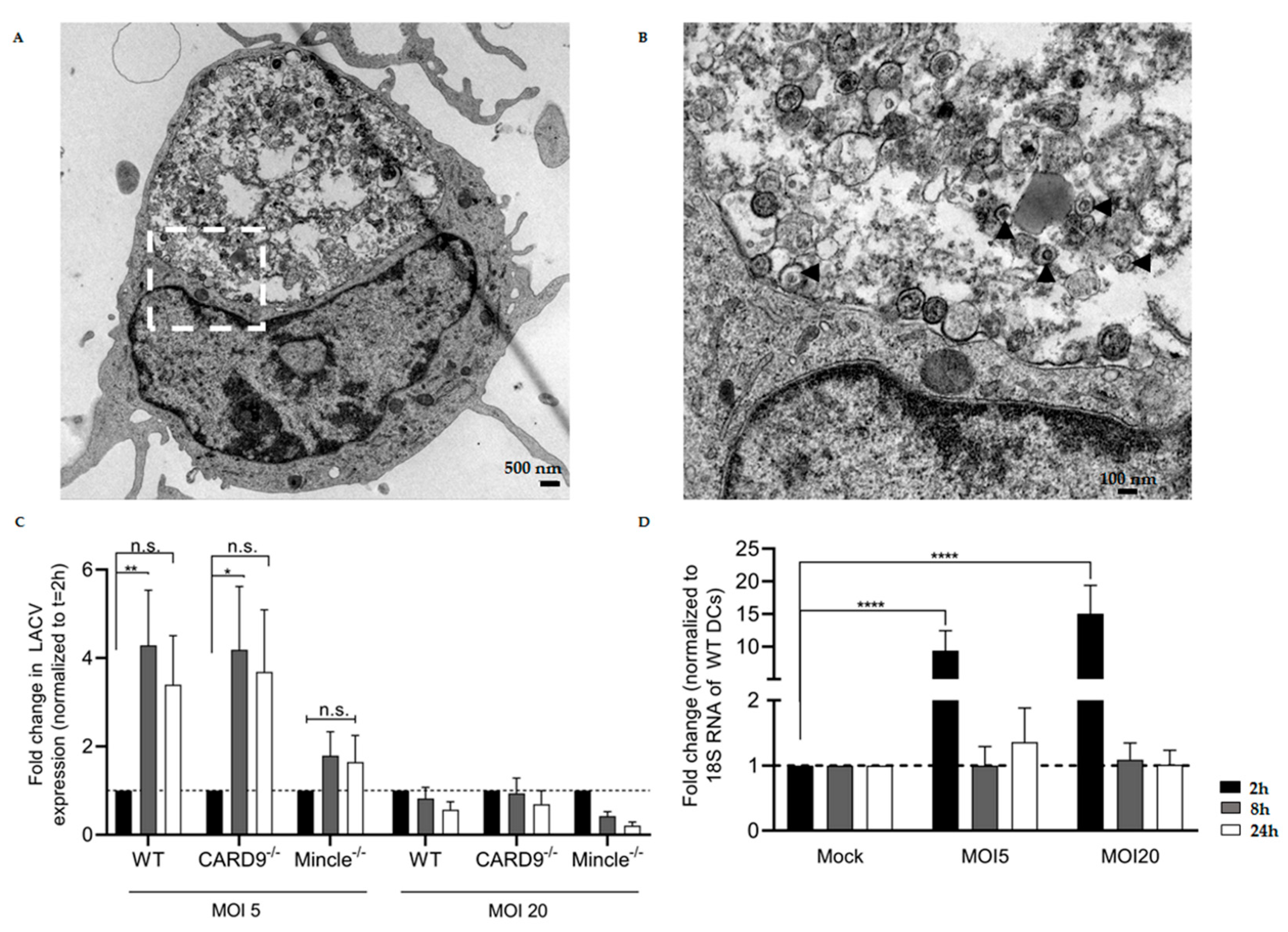
3.3. LACV Is Internalized by DCs
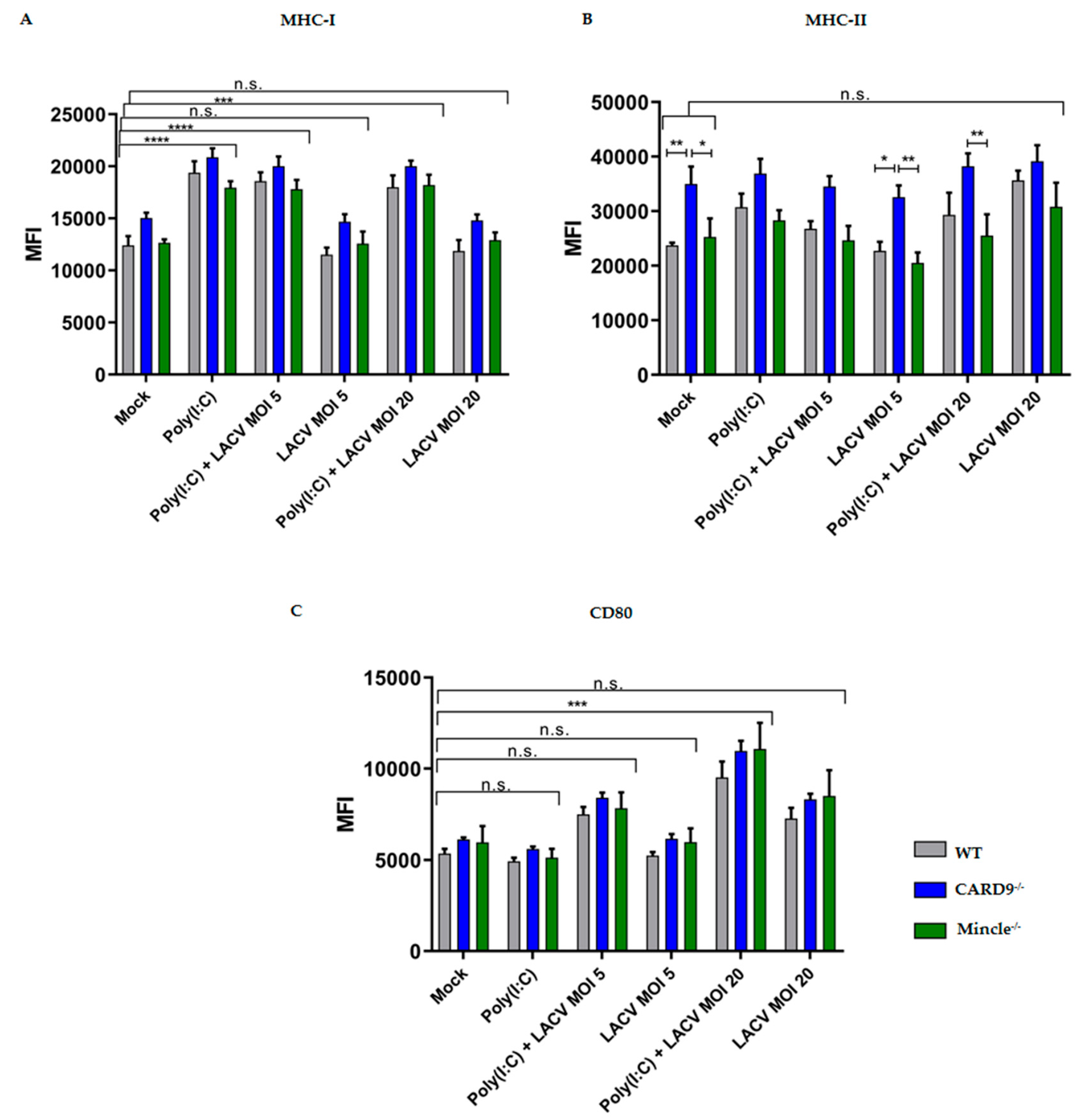
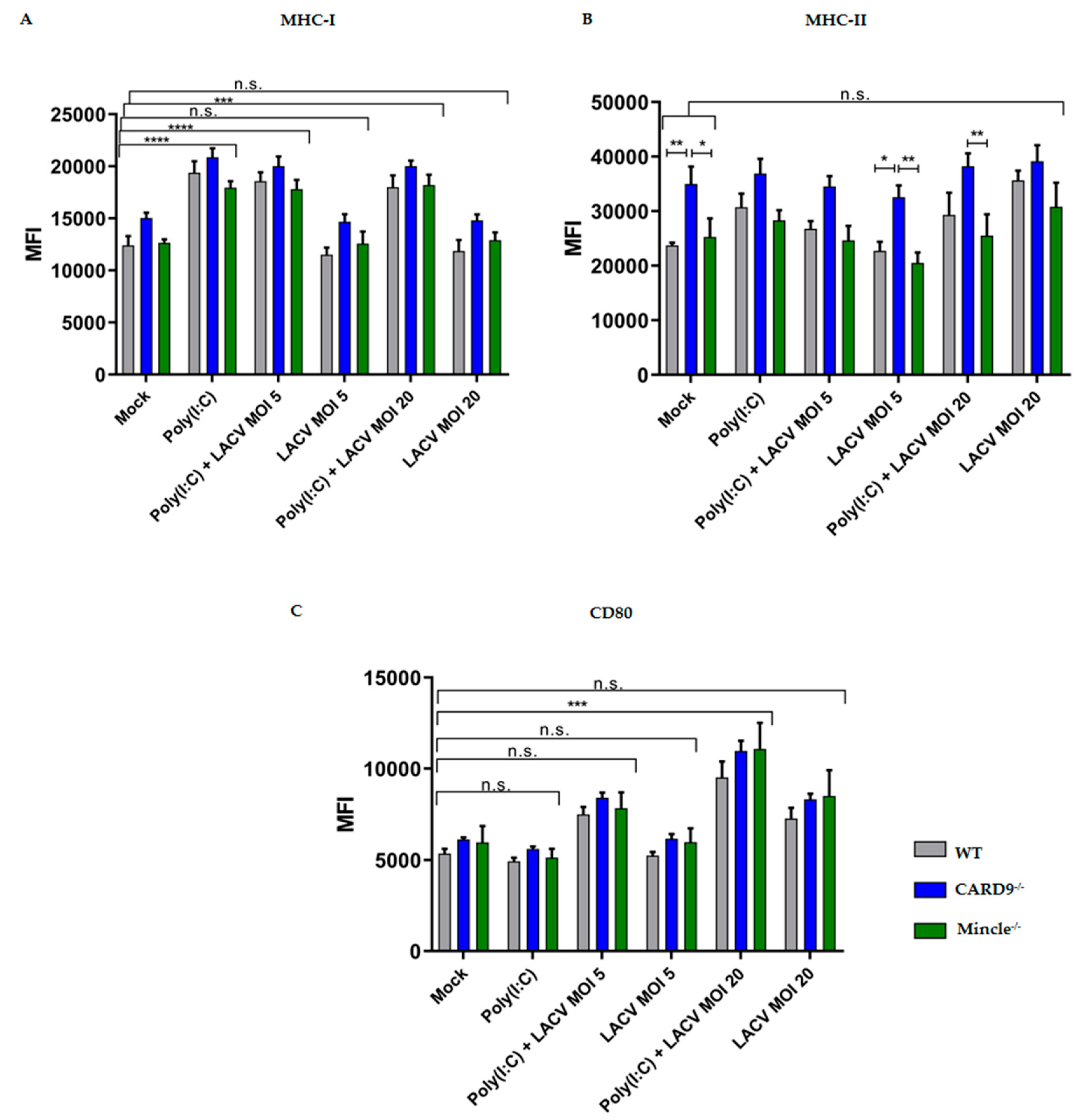
3.4. Activation of DCs by LACV
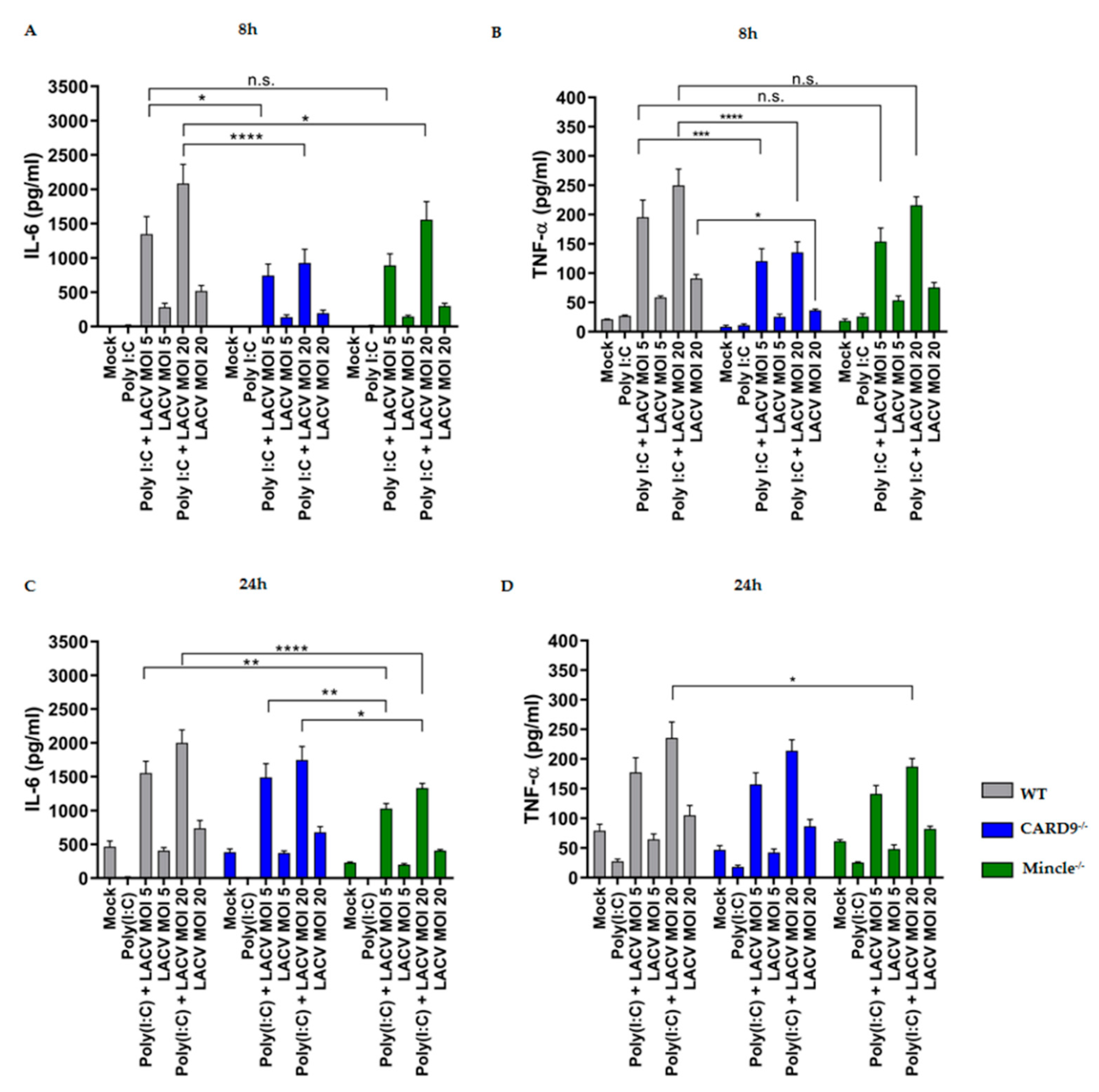
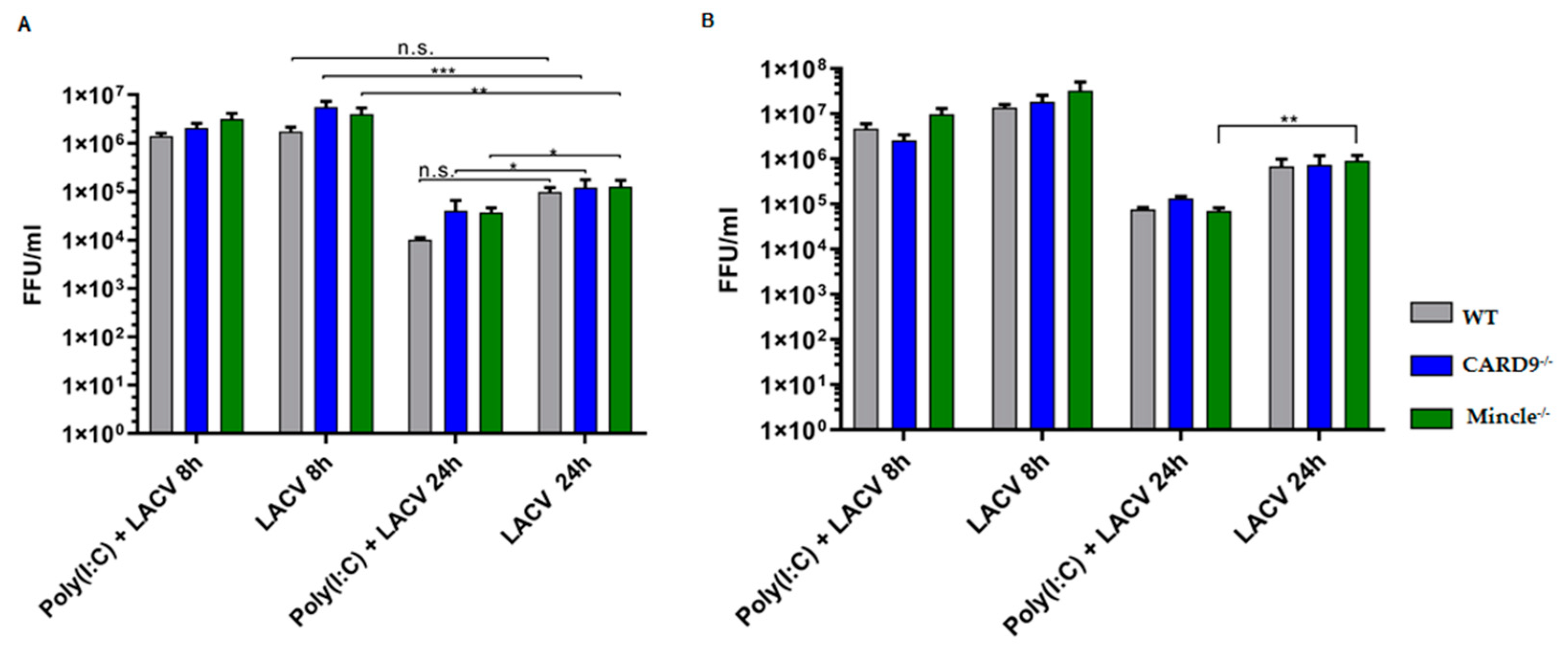
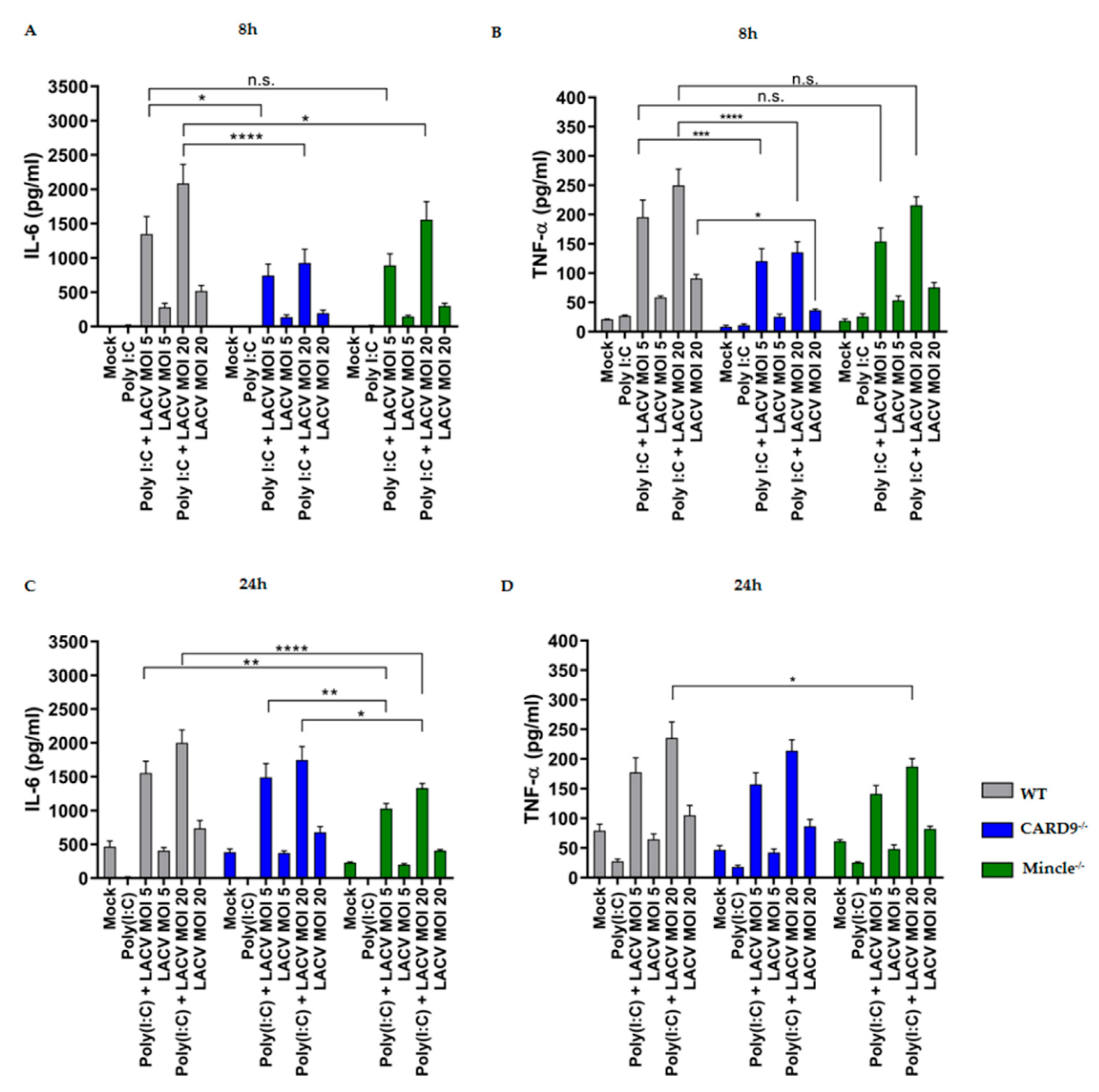
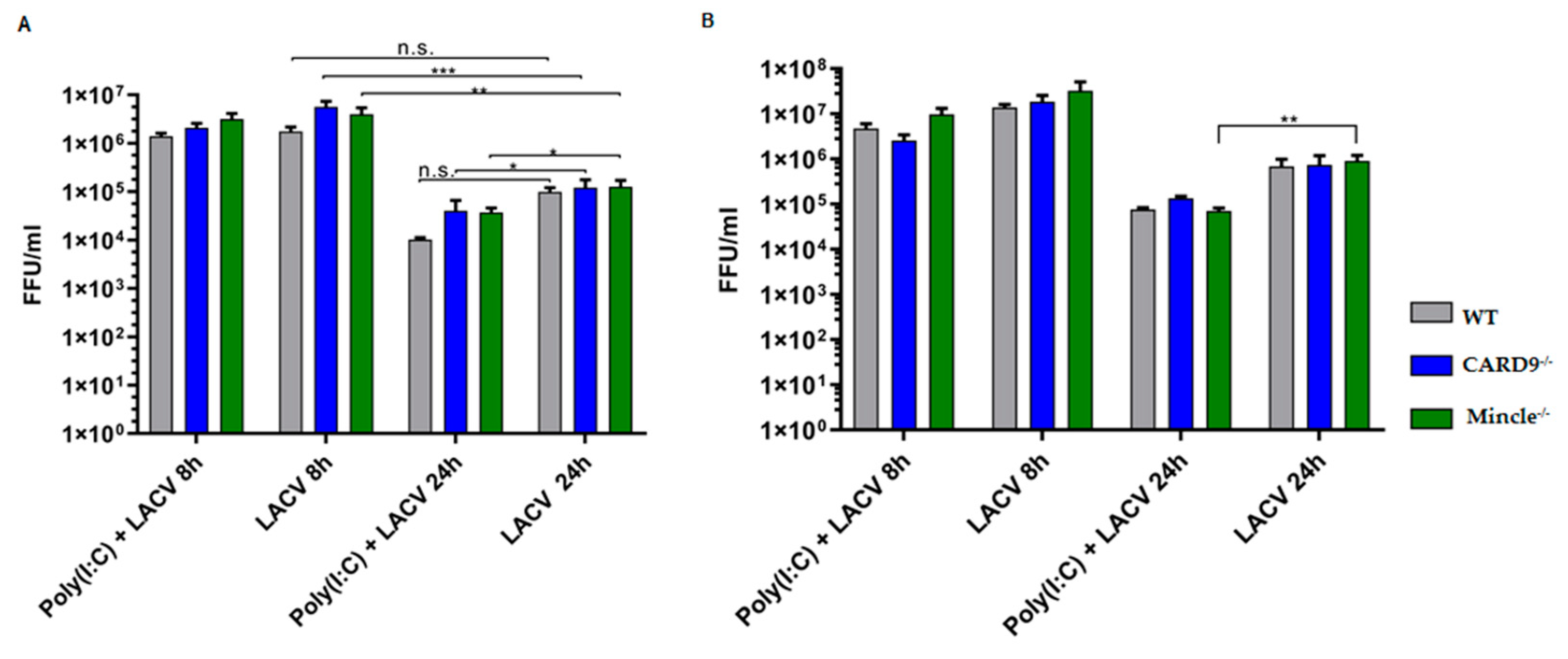
3.5. Mincle and CARD9 Contribute to Proinflammatory Signaling of LACV Infected DCs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McJunkin, J.E.; de los Reyes, E.C.; Irazuzta, J.E.; Caceres, M.J.; Khan, R.R.; Minnich, L.L.; Fu, K.D.; Lovett, G.D.; Tsai, T.; Thompson, A. La Crosse Encephalitis in Children. N. Engl. J. Med. 2001, 344, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2017). Arch. Virol. 2017, 162, 2505–2538. [Google Scholar] [CrossRef]
- Haddow, A.D.; Odoi, A. The Incidence Risk, Clustering, and Clinical Presentation of La Crosse Virus Infections in the Eastern United States, 2003–2007. PLoS ONE 2009, 4, 1–8. [Google Scholar] [CrossRef]
- Miller, A.; Carchman, R.; Long, R.; Denslow, S.A. La Crosse Viral Infection in Hospitalized Pediatric Patients in Western North Carolina. Hosp. Pediatr. 2012, 2, 235–242. [Google Scholar] [CrossRef]
- Prendergast, A.J.; Klenerman, P.; Goulder, P.J.R. The impact of differential antiviral immunity in children and adults. Nat. Rev. Immunol. 2012, 12, 636. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.K.; Hollander, G.A.; McMichael, A. Evolution of the immune system in humans from infancy to old age. Proc. Biol. Sci. 2015, 282, 20143085. [Google Scholar] [CrossRef]
- Gaensbauer, J.T.; Lindsey, N.P.; Messacar, K.; Staples, J.E.; Fischer, M. Neuroinvasive Arboviral Disease in the United States: 2003 to 2012. Pediatrics 2014, 134, e642–e650. [Google Scholar] [CrossRef]
- Haddow, A.D.; Bixler, D.; Odoi, A. The spatial epidemiology and clinical features of reported cases of La Crosse Virus infection in West Virginia from 2003 to 2007. BMC Infect. Dis. 2011, 11, 29. [Google Scholar] [CrossRef]
- Haddow, A.D.; Jones, C.J.; Odoi, A. Assessing Risk in Focal Arboviral Infections: Are We Missing the Big or Little Picture? PLoS ONE 2009, 4, e6954. [Google Scholar] [CrossRef] [PubMed]
- Albornoz, A.; Hoffmann, A.B.; Lozach, P.-Y.; Tischler, N.D. Early Bunyavirus-Host Cell Interactions. Viruses 2016, 8, 143. [Google Scholar] [CrossRef] [PubMed]
- Verbruggen, P.; Ruf, M.; Blakqori, G.; Överby, A.K.; Heidemann, M.; Eick, D.; Weber, F. Interferon antagonist NSs of La Crosse virus triggers a DNA damage response-like degradation of transcribing RNA polymerase II. J. Biol. Chem. 2011, 286, 3681–3692. [Google Scholar] [CrossRef]
- Mukherjee, P.; Woods, T.A.; Moore, R.A.; Peterson, K.E. Activation of the innate signaling molecule MAVS by bunyavirus infection upregulates the adaptor protein SARM1, leading to neuronal death. Immunity 2013, 38, 705–716. [Google Scholar] [CrossRef]
- Weber, M.; Gawanbacht, A.; Habjan, M.; Rang, A.; Borner, C.; Schmidt, A.M.; Veitinger, S.; Jacob, R.; Devignot, S.; Kochs, G.; et al. Incoming RNA virus nucleocapsids containing a 5’-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe 2013, 13, 336–346. [Google Scholar] [CrossRef]
- Taylor, K.G.; Woods, T.A.; Winkler, C.W.; Carmody, A.B.; Peterson, K.E. Age-dependent myeloid dendritic cell responses mediate resistance to la crosse virus-induced neurological disease. J. Virol. 2014, 88, 11070–11079. [Google Scholar] [CrossRef] [PubMed]
- Mayer, S.; Raulf, M.-K.; Lepenies, B. C-type lectins: Their network and roles in pathogen recognition and immunity. Histochem. Cell Biol. 2017, 147, 223–237. [Google Scholar] [CrossRef]
- Brown, G.D.; Willment, J.A.; Whitehead, L. C-type lectins in immunity and homeostasis. Nat. Rev. Immunol. 2018, 18, 374–389. [Google Scholar] [CrossRef]
- Bermejo-Jambrina, M.; Eder, J.; Helgers, L.C.; Hertoghs, N.; Nijmeijer, B.M.; Stunnenberg, M.; Geijtenbeek, T.B.H. C-Type Lectin Receptors in Antiviral Immunity and Viral Escape. Front. Immunol. 2018, 9, 590. [Google Scholar] [CrossRef]
- Monteiro, J.T.; Lepenies, B. Myeloid C-Type Lectin Receptors in Viral Recognition and Antiviral Immunity. Viruses 2017, 9, 59. [Google Scholar] [CrossRef]
- Van Breedam, W.; Pöhlmann, S.; Favoreel, H.W.; de Groot, R.J.; Nauwynck, H.J. Bitter-sweet symphony: Glycan–lectin interactions in virus biology. FEMS Microbiol. Rev. 2014, 38, 598–632. [Google Scholar] [CrossRef] [PubMed]
- Plassmeyer, M.L.; Soldan, S.S.; Stachelek, K.M.; Roth, S.M.; Martín-García, J.; González-Scarano, F. Mutagenesis of the La Crosse Virus glycoprotein supports a role for Gc (1066–1087) as the fusion peptide. Virology 2007, 358, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Brauburger, K.; Elliott, R.M. Role of N-Linked Glycans on Bunyamwera Virus Glycoproteins in Intracellular Trafficking, Protein Folding, and Virus Infectivity. J. Virol. 2005, 79, 13725–13734. [Google Scholar] [CrossRef]
- Hollidge, B.S.; Nedelsky, N.B.; Salzano, M.-V.; Fraser, J.W.; González-Scarano, F.; Soldan, S.S. Orthobunyavirus entry into neurons and other mammalian cells occurs via clathrin-mediated endocytosis and requires trafficking into early endosomes. J. Virol. 2012, 86, 7988–8001. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.; Li, X.; Zhang, X.; Liu, W.; Kühl, A.; Kaup, F.; Soldan, S.S.; González-Scarano, F.; Weber, F.; He, Y.; et al. Severe fever with thrombocytopenia virus glycoproteins are targeted by neutralizing antibodies and can use DC-SIGN as a receptor for pH-dependent entry into human and animal cell lines. J. Virol. 2013, 87, 4384–4394. [Google Scholar] [CrossRef] [PubMed]
- Lozach, P.-Y.; Kühbacher, A.; Meier, R.; Mancini, R.; Bitto, D.; Bouloy, M.; Helenius, A. DC-SIGN as a Receptor for Phleboviruses. Cell Host Microbe 2011, 10, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Teng, O.; Chen, S.-T.; Hsu, T.-L.; Sia, S.F.; Cole, S.; Valkenburg, S.A.; Hsu, T.-Y.; Zheng, J.T.; Tu, W.; Bruzzone, R.; et al. CLEC5A-mediated enhancement of the inflammatory response in myeloid cells contributes to influenza pathogenicity in vivo. J. Virol. 2016, 91, e01813-16. [Google Scholar] [CrossRef] [PubMed]
- Tani, H.; Shimojima, M.; Fukushi, S.; Yoshikawa, T.; Fukuma, A.; Taniguchi, S.; Morikawa, S.; Saijo, M. Characterization of Glycoprotein-Mediated Entry of Severe Fever with Thrombocytopenia Syndrome Virus. J. Virol. 2016, 90, 5292–5301. [Google Scholar] [CrossRef]
- Mayer, S.; Moeller, R.; Monteiro, J.T.; Ellrott, K.; Josenhans, C.; Lepenies, B. C-Type Lectin Receptor (CLR)-Fc Fusion Proteins As Tools to Screen for Novel CLR/Bacteria Interactions: An Exemplary Study on Preselected Campylobacter jejuni Isolates. Front. Immunol. 2018, 9, 213. [Google Scholar] [CrossRef] [PubMed]
- Cutts, T.; Grolla, A.; Jones, S.; Cook, B.W.M.; Qiu, X.; Theriault, S.S. Inactivation of Zaire ebolavirus Variant Makona in Human Serum Samples Analyzed by Enzyme-Linked Immunosorbent Assay. J. Infect. Dis. 2016, 214, S218–S221. [Google Scholar] [CrossRef]
- Ostrop, J.; Lang, R. Contact, Collaboration, and Conflict: Signal Integration of Syk-Coupled C-Type Lectin Receptors. J. Immunol. 2017, 198, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like Receptor and RIG-1-like Receptor Signaling. Ann. N. Y. Acad. Sci. 2008, 1143, 1–20. [Google Scholar] [CrossRef]
- Artigas, G.; Monteiro, J.T.; Hinou, H.; Nishimura, S.-I.; Lepenies, B.; Garcia-Martin, F. Glycopeptides as Targets for Dendritic Cells: Exploring MUC1 Glycopeptides Binding Profile toward Macrophage Galactose-Type Lectin (MGL) Orthologs. J. Med. Chem. 2017, 60, 9012–9021. [Google Scholar] [CrossRef]
- Stockinger, B.; Zal, T.; Zal, A.; Gray, D. B cells solicit their own help from T cells. J. Exp. Med. 1996, 183, 891–899. [Google Scholar] [CrossRef]
- Tseng, Y.-F.; Weng, T.-C.; Lai, C.-C.; Chen, P.-L.; Lee, M.-S.; Hu, A.Y.-C. A fast and efficient purification platform for cell-based influenza viruses by flow-through chromatography. Vaccine 2018, 36, 3146–3152. [Google Scholar] [CrossRef] [PubMed]
- James, K.T.; Cooney, B.; Agopsowicz, K.; Trevors, M.A.; Mohamed, A.; Stoltz, D.; Hitt, M.; Shmulevitz, M. Novel High-throughput Approach for Purification of Infectious Virions. Sci. Rep. 2016, 6, 36826. [Google Scholar] [CrossRef]
- Maglinao, M.; Eriksson, M.; Schlegel, M.K.; Zimmermann, S.; Johannssen, T.; Götze, S.; Seeberger, P.H.; Lepenies, B. A platform to screen for C-type lectin receptor-binding carbohydrates and their potential for cell-specific targeting and immune modulation. J. Control. Release 2014, 175, 36–42. [Google Scholar] [CrossRef]
- Wang, H.; Nattanmai, S.; Kramer, L.D.; Bernard, K.A.; Tavakoli, N.P. A duplex real-time reverse transcriptase polymerase chain reaction assay for the detection of California serogroup and Cache Valley viruses. Diagn. Microbiol. Infect. Dis. 2009, 65, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-G.; Siripanyaphinyo, U.; Tumkosit, U.; Noranate, N.; A-Nuegoonpipat, A.; Pan, Y.; Kameoka, M.; Kurosu, T.; Ikuta, K.; Takeda, N.; et al. Poly (I:C), an agonist of toll-like receptor-3, inhibits replication of the Chikungunya virus in BEAS-2B cells. Virol. J. 2012, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Murphy, F.; Gibbs, E.; Horzinek, M.; Michael, S. Veterinary Virology, 3rd ed.; Academic Press: Cambridge, MA, USA, 1999; ISBN 9780080552033. [Google Scholar]
- Gerhauser, I.; Li, L.; Li, D.; Klein, S.; Elmarabet, S.A.; Deschl, U.; Kalkuhl, A.; Baumgärtner, W.; Ulrich, R.; Beineke, A. Dynamic changes and molecular analysis of cell death in the spinal cord of SJL mice infected with the BeAn strain of Theiler’s murine encephalomyelitis virus. Apoptosis 2018, 23, 170–186. [Google Scholar] [CrossRef]
- Kummerfeld, M.; Meens, J.; Haas, L.; Baumgärtner, W.; Beineke, A. Generation and characterization of a polyclonal antibody for the detection of Theiler’s murine encephalomyelitis virus by light and electron microscopy. J. Virol. Methods 2009, 160, 185–188. [Google Scholar] [CrossRef]
- Chen, S.-T.; Lin, Y.-L.; Huang, M.-T.; Wu, M.-F.; Cheng, S.-C.; Lei, H.-Y.; Lee, C.-K.; Chiou, T.-W.; Wong, C.-H.; Hsieh, S.-L. CLEC5A is critical for dengue-virus-induced lethal disease. Nature 2008, 453, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Van der Vlist, M.; Geijtenbeek, T.B.H. Langerin functions as an antiviral receptor on Langerhans cells. Immunol. Cell Biol. 2010, 88, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Léger, P.; Tetard, M.; Youness, B.; Cordes, N.; Rouxel, R.N.; Flamand, M.; Lozach, P.-Y. Differential Use of the C-Type Lectins L-SIGN and DC-SIGN for Phlebovirus Endocytosis. Traffic 2016, 17, 639–656. [Google Scholar] [CrossRef]
- Pérez de Diego, R.; Sánchez-Ramón, S.; López-Collazo, E.; Martínez-Barricarte, R.; Cubillos-Zapata, C.; Ferreira Cerdán, A.; Casanova, J.-L.; Puel, A. Genetic errors of the human caspase recruitment domain-B-cell lymphoma 10-mucosa-associated lymphoid tissue lymphoma-translocation gene 1 (CBM) complex: Molecular, immunologic, and clinical heterogeneity. J. Allergy Clin. Immunol. 2015, 136, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Levine, S.J. Toll-like receptor, RIG-I-like receptors and the NLRP3 inflammasome: Key modulators of innate immune responses to double-stranded RNA viruses. Cytokine Growth Factor Rev. 2011, 22, 63–72. [Google Scholar] [CrossRef]
- Tan, R.S.T.; Ho, B.; Leung, B.P.; Ding, J.L. TLR cross-talk confers specificity to innate immunity. Int. Rev. Immunol. 2014, 33, 443–453. [Google Scholar] [CrossRef]
- Hefti, H.P.; Frese, M.; Landis, H.; Di Paolo, C.; Aguzzi, A.; Haller, O.; Pavlovic, J. Human MxA Protein Protects Mice Lacking a Functional Alpha/Beta Interferon System against La Crosse Virus and Other Lethal Viral Infections. J. Virol. 1999, 73, 6984–6991. [Google Scholar]
- Pavlovic, J.; Schultz, J.; Hefti, H.P.; Schuh, T.; Mölling, K. DNA Vaccination against La Crosse Virus. Intervirology 2000, 43, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Slater, L.; Bartlett, N.W.; Haas, J.J.; Zhu, J.; Message, S.D.; Walton, R.P.; Sykes, A.; Dahdaleh, S.; Clarke, D.L.; Belvisi, M.G.; et al. Co-ordinated Role of TLR3, RIG-I and MDA5 in the Innate Response to Rhinovirus in Bronchial Epithelium. PLoS Pathog. 2010, 6, e1001178. [Google Scholar] [CrossRef] [PubMed]
- Gibbert, K.; Dietze, K.K.; Zelinskyy, G.; Lang, K.S.; Barchet, W.; Kirschning, C.J.; Dittmer, U. Polyinosinic-Polycytidylic Acid Treatment of Friend Retrovirus-Infected Mice Improves Functional Properties of Virus-Specific T Cells and Prevents Virus-Induced Disease. J. Immunol. 2010, 185, 6179–6189. [Google Scholar] [CrossRef]
- Ngoi, S.M.; Tovey, M.G.; Vella, A.T. Targeting Poly(I:C) to the TLR3-Independent Pathway Boosts Effector CD8 T Cell Differentiation through IFN-α/β. J. Immunol. 2008, 181, 7670–7680. [Google Scholar] [CrossRef]
- Luby, J.P. Sensitivities of Neurotropic Arboviruses to Human Interferon. J. Infect. Dis. 1975, 132, 361–367. [Google Scholar] [CrossRef]
- Gerhardt, R.R.; Gottfried, K.L.; Apperson, C.S.; Davis, B.S.; Erwin, P.C.; Smith, A.B.; Panella, N.A.; Powell, E.E.; Nasci, R.S. First isolation of La Crosse virus from naturally infected Aedes albopictus. Emerg. Infect. Dis. 2001, 7, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.C.; Yang, F.; Jackson, D.M.; Dotseth, E.J.; Paulson, S.L.; Hawley, D.M. La Crosse Virus Field Detection and Vector Competence of Culex Mosquitoes. Am. J. Trop. Med. Hyg. 2015, 93, 461–467. [Google Scholar] [CrossRef]
- Sancho, D.; Reis e Sousa, C. Sensing of cell death by myeloid C-type lectin receptors. Curr. Opin. Immunol. 2013, 25, 46–52. [Google Scholar] [CrossRef]
- Hanč, P.; Fujii, T.; Iborra, S.; Yamada, Y.; Huotari, J.; Schulz, O.; Ahrens, S.; Kjær, S.; Way, M.; Sancho, D.; et al. Structure of the Complex of F-Actin and DNGR-1, a C-Type Lectin Receptor Involved in Dendritic Cell Cross-Presentation of Dead Cell-Associated Antigens. Immunity 2015, 42, 839–849. [Google Scholar] [CrossRef]
- Neumann, K.; Castiñeiras-Vilariño, M.; Höckendorf, U.; Hannesschläger, N.; Lemeer, S.; Kupka, D.; Meyermann, S.; Lech, M.; Anders, H.-J.; Kuster, B.; et al. Clec12a Is an Inhibitory Receptor for Uric Acid Crystals that Regulates Inflammation in Response to Cell Death. Immunity 2014, 40, 389–399. [Google Scholar] [CrossRef]
- Yuita, H.; Tsuiji, M.; Tajika, Y.; Matsumoto, Y.; Hirano, K.; Suzuki, N.; Irimura, T. Retardation of removal of radiation-induced apoptotic cells in developing neural tubes in macrophage galactose-type C-type lectin-1-deficient mouse embryos. Glycobiology 2005, 15, 1368–1375. [Google Scholar] [CrossRef]
- Yamasaki, S.; Ishikawa, E.; Sakuma, M.; Hara, H.; Ogata, K.; Saito, T. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat. Immunol. 2008, 9, 1179. [Google Scholar] [CrossRef]
- Kiyotake, R.; Oh-hora, M.; Ishikawa, E.; Miyamoto, T.; Ishibashi, T.; Yamasaki, S. Human Mincle Binds to Cholesterol Crystals and Triggers Innate Immune Responses. J. Biol. Chem. 2015, 290, 25322–25332. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M.; Izumi, Y.; Ishikawa, E.; Kiyotake, R.; Doi, R.; Iwai, S.; Omahdi, Z.; Yamaji, T.; Miyamoto, T.; Bamba, T.; et al. Intracellular metabolite β-glucosylceramide is an endogenous Mincle ligand possessing immunostimulatory activity. Proc. Natl. Acad. Sci. USA 2017, 114, E3285–E3294. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.C.; Londrigan, S.L.; Nasr, N.; Cunningham, A.L.; Turville, S.; Brooks, A.G.; Reading, P.C. The C-type lectin langerin functions as a receptor for attachment and infectious entry of influenza A virus. J. Virol. 2015, 90, 206–221. [Google Scholar] [CrossRef]
- Usami, K.; Matsuno, K.; Igarashi, M.; Denda-Nagai, K.; Takada, A.; Irimura, T. Involvement of viral envelope GP2 in Ebola virus entry into cells expressing the macrophage galactose-type C-type lectin. Biochem. Biophys. Res. Commun. 2011, 407, 74–78. [Google Scholar] [CrossRef]
- Ng, W.C.; Liong, S.; Tate, M.D.; Irimura, T.; Denda-Nagai, K.; Brooks, A.G.; Londrigan, S.L.; Reading, P.C. The Macrophage Galactose-Type Lectin Can Function as an Attachment and Entry Receptor for Influenza Virus. J. Virol. 2014, 88, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Matsumura, T.; Mayer-Lambertz, S.; Wells, C.A.; Ishikawa, E.; Butcher, S.K.; Barnett, T.C.; Walker, M.J.; Imamura, A.; Ishida, H.; et al. Lipoteichoic acid anchor triggers Mincle to drive protective immunity against invasive group A Streptococcus infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10662–E10671. [Google Scholar] [CrossRef] [PubMed]
- Wevers, B.A.; Kaptein, T.M.; Zijlstra-Willems, E.M.; Theelen, B.; Boekhout, T.; Geijtenbeek, T.B.H.; Gringhuis, S.I. Fungal Engagement of the C-Type Lectin Mincle Suppresses Dectin-1-Induced Antifungal Immunity. Cell Host Microbe 2014, 15, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Nagata, M.; Yamasaki, S. Mincle: 20 years of a versatile sensor of insults. Int. Immunol. 2018, 30, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Decout, A.; Silva-Gomes, S.; Drocourt, D.; Blattes, E.; Rivière, M.; Prandi, J.; Larrouy-Maumus, G.; Caminade, A.-M.; Hamasur, B.; Källenius, G.; et al. Deciphering the molecular basis of mycobacteria and lipoglycan recognition by the C-type lectin Dectin-2. Sci. Rep. 2018, 8, 16840. [Google Scholar] [CrossRef]
- Goodridge, H.S.; Reyes, C.N.; Becker, C.A.; Katsumoto, T.R.; Ma, J.; Wolf, A.J.; Bose, N.; Chan, A.S.H.; Magee, A.S.; Danielson, M.E.; et al. Activation of the innate immune receptor Dectin-1 upon formation of a “phagocytic synapse”. Nature 2011, 472, 471–475. [Google Scholar] [CrossRef]
- Saijo, S.; Iwakura, Y. Dectin-1 and Dectin-2 in innate immunity against fungi. Int. Immunol. 2011, 23, 467–472. [Google Scholar] [CrossRef]
- Yamasaki, S.; Matsumoto, M.; Takeuchi, O.; Matsuzawa, T.; Ishikawa, E.; Sakuma, M.; Tateno, H.; Uno, J.; Hirabayashi, J.; Mikami, Y.; et al. C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc. Natl. Acad. Sci. USA 2009, 106, 1897–1902. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, E.; Ishikawa, T.; Morita, Y.S.; Toyonaga, K.; Yamada, H.; Takeuchi, O.; Kinoshita, T.; Akira, S.; Yoshikai, Y.; Yamasaki, S. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J. Exp. Med. 2009, 206, 2879–2888. [Google Scholar] [CrossRef]
- Poeck, H.; Bscheider, M.; Gross, O.; Finger, K.; Roth, S.; Rebsamen, M.; Hannesschläger, N.; Schlee, M.; Rothenfusser, S.; Barchet, W.; et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1β production. Nat. Immunol. 2009, 11, 63. [Google Scholar] [CrossRef]
- Del Fresno, C.; Soulat, D.; Roth, S.; Blazek, K.; Udalova, I.; Sancho, D.; Ruland, J.; Ardavín, C. Interferon-β Production via Dectin-1-Syk-IRF5 Signaling in Dendritic Cells Is Crucial for Immunity to C. albicans. Immunity 2013, 38, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.M.; Bidle, K.D. Attenuation of virus production at high multiplicities of infection in Aureococcus anophagefferens. Virology 2014, 466–467, 71–81. [Google Scholar] [CrossRef]
- Rüdiger, D.; Kupke, S.Y.; Laske, T.; Zmora, P.; Reichl, U. Multiscale modeling of influenza A virus replication in cell cultures predicts infection dynamics for highly different infection conditions. PLoS Comput. Biol. 2019, 15, 1–22. [Google Scholar] [CrossRef]
- Akpinar, F.; Inankur, B.; Yin, J. Spatial-Temporal Patterns of Viral Amplification and Interference Initiated by a Single Infected Cell. J. Virol. 2016, 90, 7552. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, T.; Iizasa, E.; Kobayashi, N.; Yoshida, H.; Hara, H. Loss of CARD9-mediated innate activation attenuates severe influenza pneumonia without compromising host viral immunity. Sci. Rep. 2015, 5, 17577. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Volume (mL) | Protein (µg/mL) | Total Protein (µg) | FFU/mL | Protein Removal (%) |
|---|---|---|---|---|---|
| LACV (initial) | 100 | 2600 ± 1200 | 260,000 | 2.3 ± 1.8 × 109 | - |
| LACV (final) | 3 | 17 ± 5.5 | 51 | 4.3 ± 1.5 × 107 | 99.4 |
| Mock (initial) | 100 | 2450 ± 1100 | 245,000 | 0 | - |
| Mock (final) | 3 | 8.5 ± 0.2 | 25.5 | 0 | 99.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monteiro, J.T.; Schön, K.; Ebbecke, T.; Goethe, R.; Ruland, J.; Baumgärtner, W.; Becker, S.C.; Lepenies, B. The CARD9-Associated C-Type Lectin, Mincle, Recognizes La Crosse Virus (LACV) but Plays a Limited Role in Early Antiviral Responses against LACV. Viruses 2019, 11, 303. https://doi.org/10.3390/v11030303
Monteiro JT, Schön K, Ebbecke T, Goethe R, Ruland J, Baumgärtner W, Becker SC, Lepenies B. The CARD9-Associated C-Type Lectin, Mincle, Recognizes La Crosse Virus (LACV) but Plays a Limited Role in Early Antiviral Responses against LACV. Viruses. 2019; 11(3):303. https://doi.org/10.3390/v11030303
Chicago/Turabian StyleMonteiro, João T., Kathleen Schön, Tim Ebbecke, Ralph Goethe, Jürgen Ruland, Wolfgang Baumgärtner, Stefanie C. Becker, and Bernd Lepenies. 2019. "The CARD9-Associated C-Type Lectin, Mincle, Recognizes La Crosse Virus (LACV) but Plays a Limited Role in Early Antiviral Responses against LACV" Viruses 11, no. 3: 303. https://doi.org/10.3390/v11030303
APA StyleMonteiro, J. T., Schön, K., Ebbecke, T., Goethe, R., Ruland, J., Baumgärtner, W., Becker, S. C., & Lepenies, B. (2019). The CARD9-Associated C-Type Lectin, Mincle, Recognizes La Crosse Virus (LACV) but Plays a Limited Role in Early Antiviral Responses against LACV. Viruses, 11(3), 303. https://doi.org/10.3390/v11030303