A Novel Hepacivirus in Wild Rodents from South America

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Rodent Samples and Ethical Statements
2.2. Viral RNA Extraction, Sequencing, and Assembly
2.3. Genome Characterization
2.4. Phylogenetic Analysis
2.5. p-Distance Analysis
2.6. Recombination Analysis
2.7. RT-PCR for Novel Hepacivirus
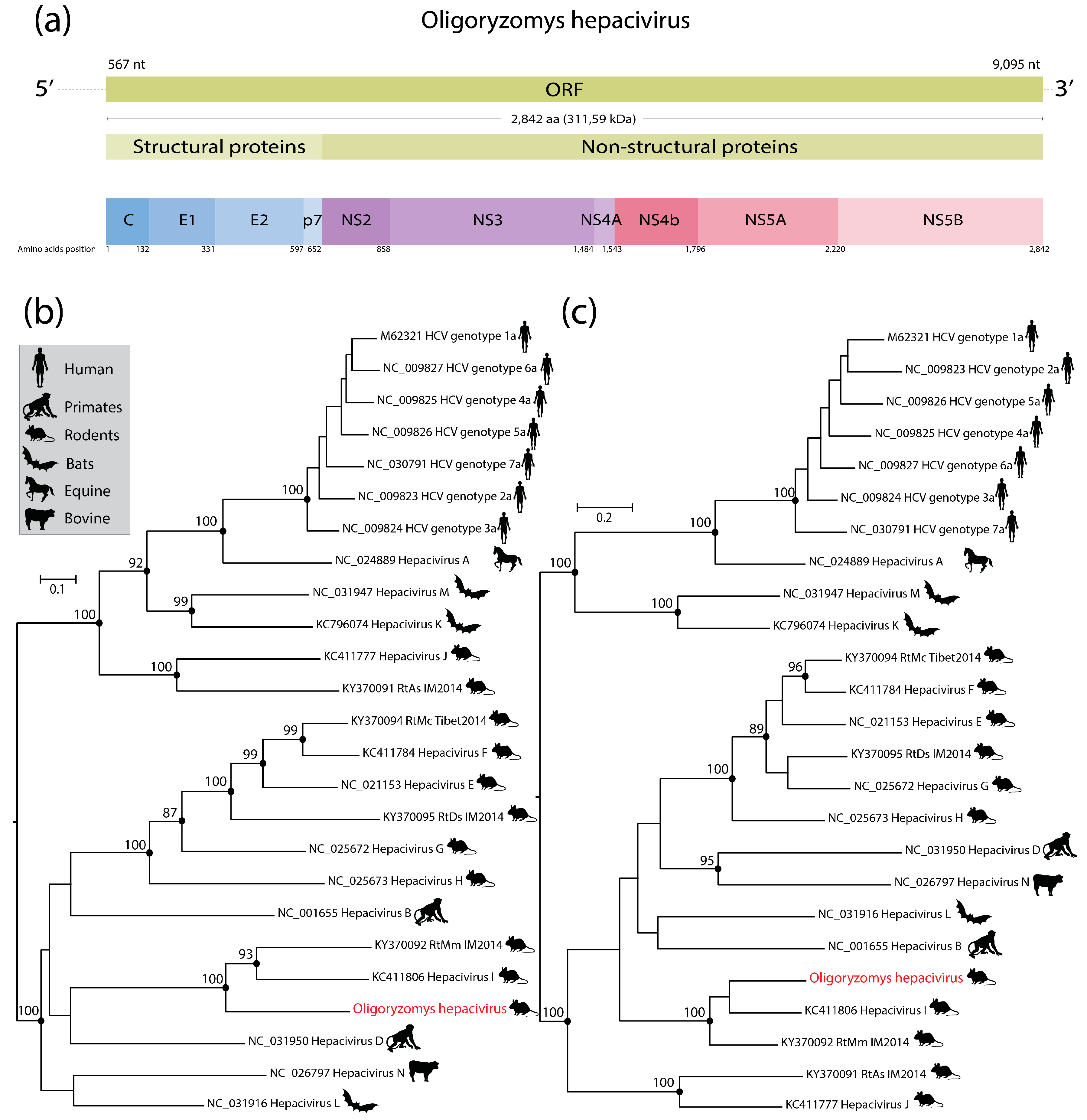
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV virus taxonomy profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Hartlage, A.S.; Cullen, J.M.; Kapoor, A. The strange, expanding world of animal hepaciviruses. Annu. Rev. Virol. 2016, 3, 53–75. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Simmonds, P.; Scheel, T.K.; Hjelle, B.; Cullen, J.M.; Burbelo, P.D.; Chauhan, L.V.; Duraisamy, R.; Sanchez Leon, M.; Jain, K.; et al. Identification of rodent homologs of hepatitis C virus and pegiviruses. mBio 2013, 4, e00216-00213. [Google Scholar] [CrossRef]
- Van Nguyen, D.; Van Nguyen, C.; Bonsall, D.; Ngo, T.T.; Carrique-Mas, J.; Pham, A.H.; Bryant, J.E.; Thwaites, G.; Baker, S.; Woolhouse, M.; et al. Detection and characterization of homologues of human hepatitis viruses and pegiviruses in rodents and bats in vietnam. Viruses 2018, 10. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Muller, M.A.; Lukashev, A.N.; Gmyl, A.; Coutard, B.; Adam, A.; Ritz, D.; Leijten, L.M.; van Riel, D.; et al. Evidence for novel hepaciviruses in rodents. PLoS Pathog. 2013, 9, e1003438. [Google Scholar] [CrossRef]
- Pfaender, S.; Brown, R.J.; Pietschmann, T.; Steinmann, E. Natural reservoirs for homologs of hepatitis C virus. Emerg. Microbes Infect. 2014, 3, e21. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 178. [Google Scholar] [CrossRef] [PubMed]
- De Souza, W.M.; Romeiro, M.F.; Sabino-Santos, G., Jr.; Maia, F.G.M.; Fumagalli, M.J.; Modha, S.; Nunes, M.R.T.; Murcia, P.R.; Figueiredo, L.T.M. Novel orthohepeviruses in wild rodents from Sao Paulo State, Brazil. Virology 2018, 519, 12–16. [Google Scholar] [CrossRef] [PubMed]
- De Souza, W.M.; Fumagalli, M.J.; de Araujo, J.; Sabino-Santos, G., Jr.; Maia, F.G.M.; Romeiro, M.F.; Modha, S.; Nardi, M.S.; Queiroz, L.H.; Durigon, E.L.; et al. Discovery of novel anelloviruses in small mammals expands the host range and diversity of the anelloviridae. Virology 2018, 514, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Souza, W.M.; Fumagalli, M.J.; Torres Carrasco, A.O.; Romeiro, M.F.; Modha, S.; Seki, M.C.; Gheller, J.M.; Daffre, S.; Nunes, M.R.T.; Murcia, P.R.; et al. Viral diversity of rhipicephalus microplus parasitizing cattle in Southern Brazil. Sci. Rep. 2018, 8, 16315. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.Y.; Dosztanyi, Z.; El-Gebali, S.; Fraser, M.; et al. Interpro in 2017-beyond protein family and domain annotations. Nucleic acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef]
- Smith, D.B.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, A.S.; Pletnev, A.; Rico-Hesse, R.; Stapleton, J.T.; et al. Proposed update to the taxonomy of the genera hepacivirus and pegivirus within the flaviviridae family. J. Gen. Virol. 2016, 97, 2894–2907. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Grishin, N.V. Promals3d: Multiple protein sequence alignment enhanced with evolutionary and three-dimensional structural information. Methods Mol. Biol. 2014, 1079, 263–271. [Google Scholar]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. Iq-tree: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. Modelfinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. Rdp4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Quan, P.L.; Firth, C.; Conte, J.M.; Williams, S.H.; Zambrana-Torrelio, C.M.; Anthony, S.J.; Ellison, J.A.; Gilbert, A.T.; Kuzmin, I.V.; Niezgoda, M.; et al. Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proc. Natl. Acad. Sci. USA 2013, 110, 8194–8199. [Google Scholar] [CrossRef]
- Billerbeck, E.; Wolfisberg, R.; Fahnoe, U.; Xiao, J.W.; Quirk, C.; Luna, J.M.; Cullen, J.M.; Hartlage, A.S.; Chiriboga, L.; Ghoshal, K.; et al. Mouse models of acute and chronic hepacivirus infection. Science 2017, 357, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Burbelo, P.D.; Dubovi, E.J.; Simmonds, P.; Medina, J.L.; Henriquez, J.A.; Mishra, N.; Wagner, J.; Tokarz, R.; Cullen, J.M.; Iadarola, M.J.; et al. Serology-enabled discovery of genetically diverse hepaciviruses in a new host. J. Virol. 2012, 86, 6171–6178. [Google Scholar] [CrossRef] [PubMed]
- Musser, G.; Carleton, M. Superfamily muroidea. In Mammal Species of the World; Wilson, D., Reeder, D., Eds.; The Johns Hopkins University Press: Baltimore, MD, USA, 2005. [Google Scholar]



{kind=link}
{kind=link}
| Pool | Species | N1 | Collection Date | Number of Reads |
|---|---|---|---|---|
| 1 | Akodon montensis | 55 | 2008 | 27,569,342 |
| 2 | Akodon montensis | 55 | 2008 | 23,439,326 |
| 3 | Akodon montensis | 41 | 2009 | 16,698,848 |
| 4 | Akodon montensis | 48 | 2012–2013 | 18,783,944 |
| 5 | Calomys tener | 38 | 2008 | 27,017,352 |
| 6 | Calomys tener | 37 | 2008 | 23,972,304 |
| 7 | Calomys tener | 34 | 2009, 2012–2013 | 15,679,756 |
| 8 | Necromys lasiurus | 59 | 2008 | 22,989,548 |
| 9 | Necromys lasiurus | 59 | 2008 | 9,252,300 |
| 10 | Necromys lasiurus | 58 | 2008 | 18,213,066 |
| 11 | Necromys lasiurus | 52 | 2009 | 22,210,888 |
| 12 | Necromys lasiurus | 24 | 2012–2013 | 25,122,228 |
| 13 | Oligoryzomys nigripes | 43 | 2008–2009 | 15,813,430 |
| 14 | Oligoryzomys nigripes | 18 | 2012–2013 | 24,796,054 |
| 15 | Mus musculus | 24 | 2008–2009 | 16,661,928 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, W.M.; Fumagalli, M.J.; Sabino-Santos, G., Jr.; Motta Maia, F.G.; Modha, S.; Teixeira Nunes, M.R.; Murcia, P.R.; Moraes Figueiredo, L.T. A Novel Hepacivirus in Wild Rodents from South America. Viruses 2019, 11, 297. https://doi.org/10.3390/v11030297
de Souza WM, Fumagalli MJ, Sabino-Santos G Jr., Motta Maia FG, Modha S, Teixeira Nunes MR, Murcia PR, Moraes Figueiredo LT. A Novel Hepacivirus in Wild Rodents from South America. Viruses. 2019; 11(3):297. https://doi.org/10.3390/v11030297
Chicago/Turabian Stylede Souza, William Marciel, Marcílio Jorge Fumagalli, Gilberto Sabino-Santos, Jr., Felipe Gonçalves Motta Maia, Sejal Modha, Márcio Roberto Teixeira Nunes, Pablo Ramiro Murcia, and Luiz Tadeu Moraes Figueiredo. 2019. "A Novel Hepacivirus in Wild Rodents from South America" Viruses 11, no. 3: 297. https://doi.org/10.3390/v11030297
APA Stylede Souza, W. M., Fumagalli, M. J., Sabino-Santos, G., Jr., Motta Maia, F. G., Modha, S., Teixeira Nunes, M. R., Murcia, P. R., & Moraes Figueiredo, L. T. (2019). A Novel Hepacivirus in Wild Rodents from South America. Viruses, 11(3), 297. https://doi.org/10.3390/v11030297