Differential Innate Immune Responses Elicited by Nipah Virus and Cedar Virus Correlate with Disparate In Vivo Pathogenesis in Hamsters


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
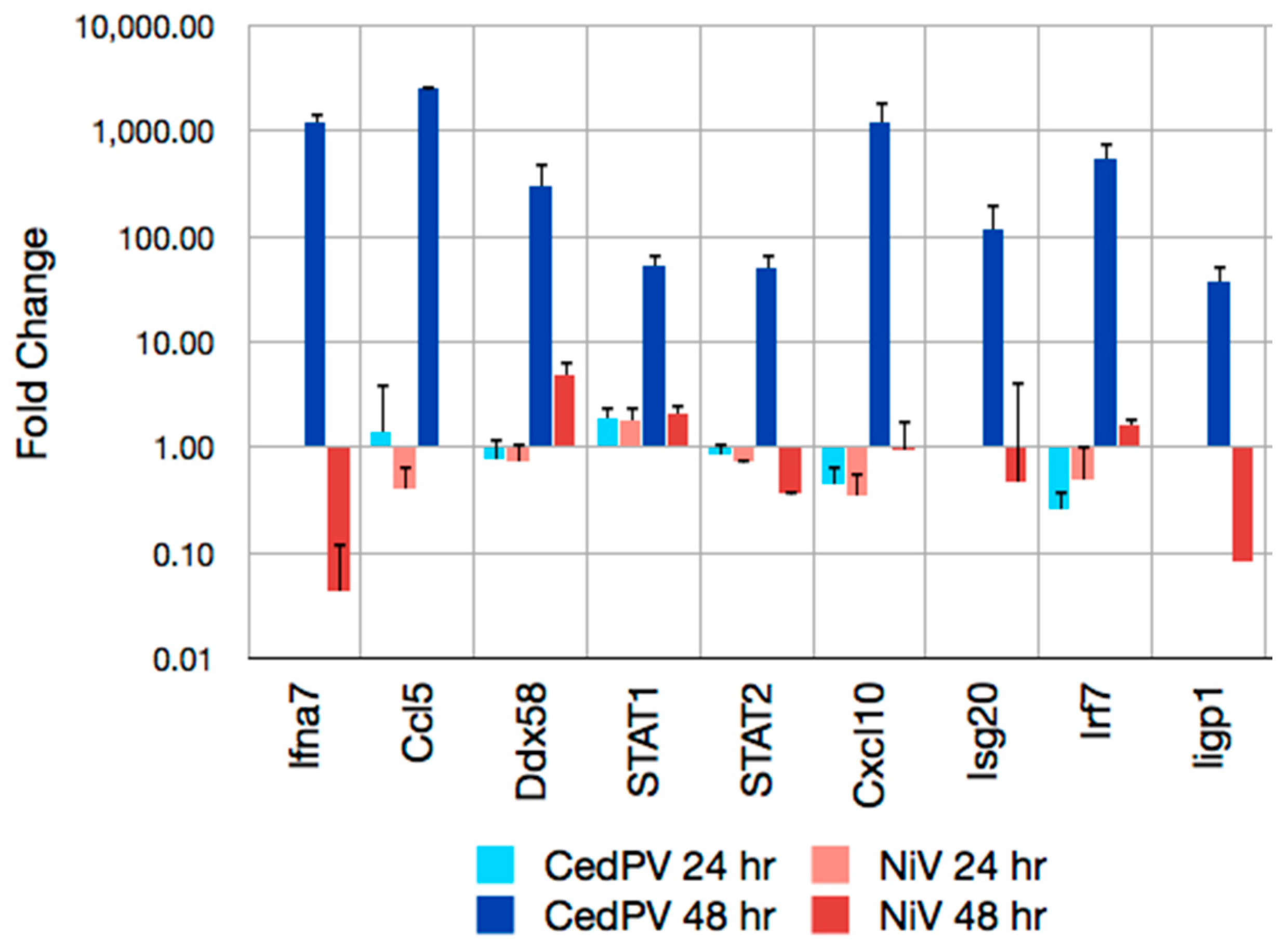{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Viruses and Cells
2.3. Hamster Infections
2.4. Virus Quantification
2.5. Virus Neutralization Assay
2.6. Generation of Primary Hamster Pulmonary Microvascular Endothelial Cells
2.7. Real-Time PCR of Host Response Genes
3. Results
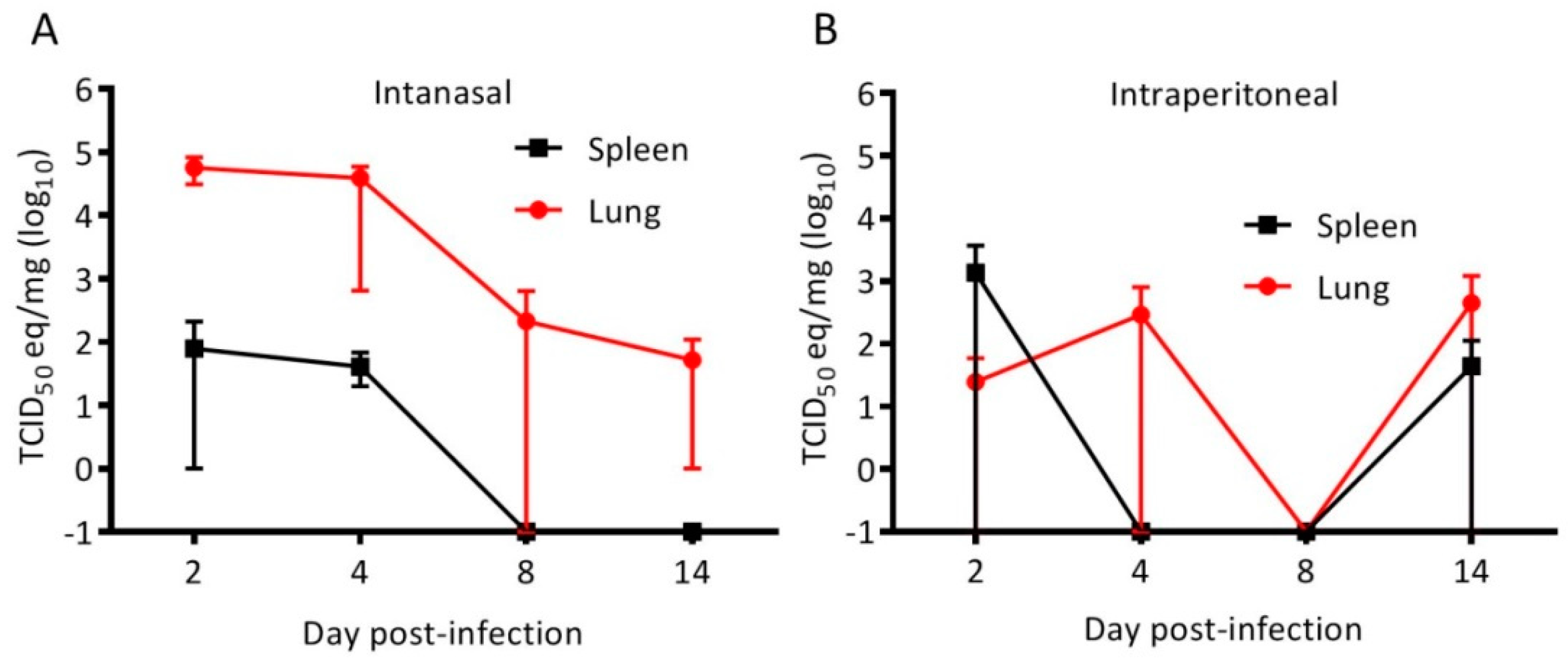
3.1. Cedar Virus Infects Hamsters without Signs of Disesae
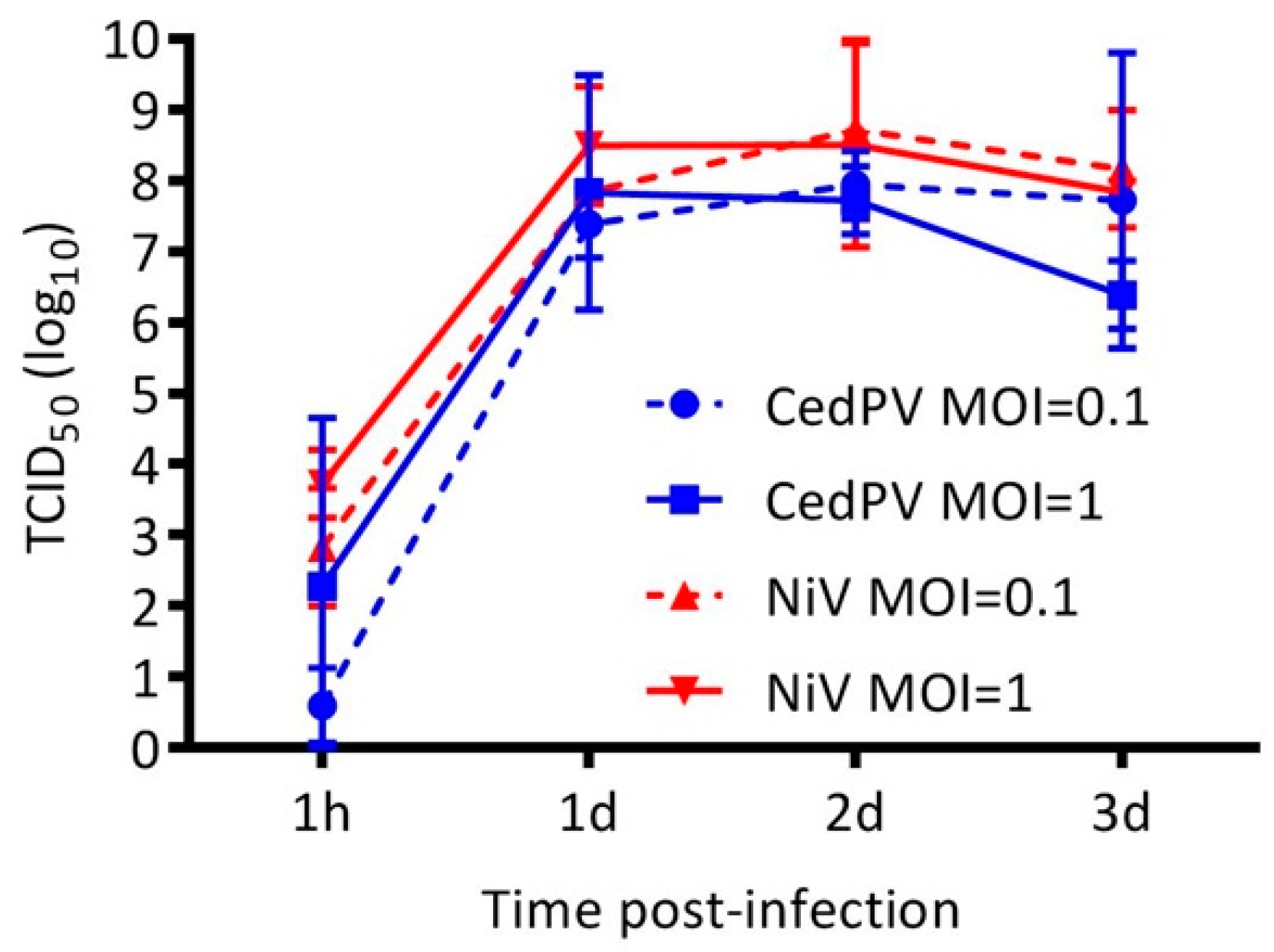
3.2. Cedar Virus Replicates in Hamster Cells
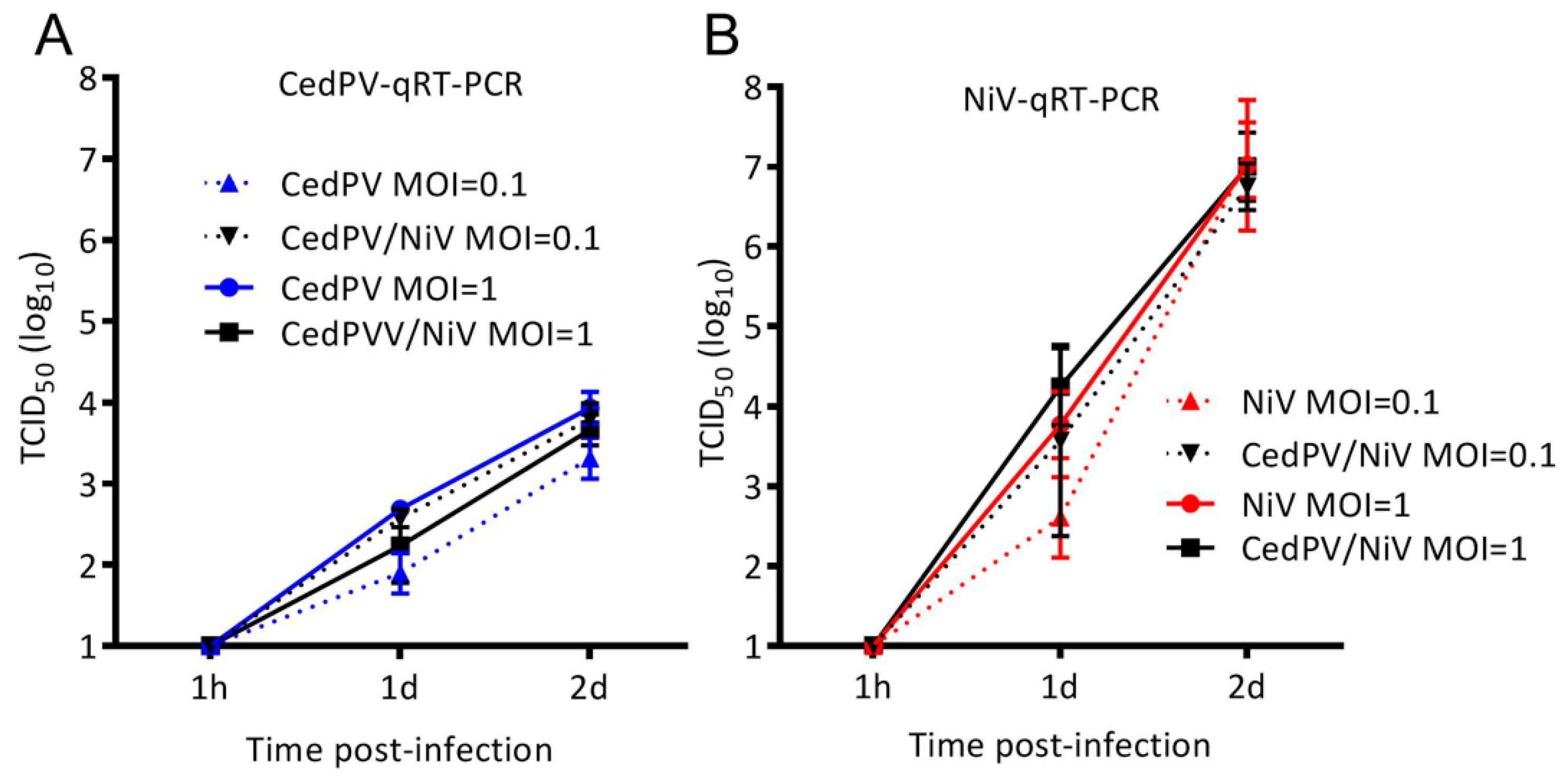
3.3. NiV Fails to Rescue CedPV Replication in Primary Hamster Endothelial Cells
3.4. CedPV Fails to Block Antiviral Defenses in Primary Hamster Endothelial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Yob, J.M.; Field, H.; Rashdi, A.M.; Morrissy, C.; van der Heide, B.; Rota, P.; bin Adzhar, A.; White, J.; Daniels, P.; Jamaluddin, A.; et al. Nipah virus infection in bats (order Chiroptera) in peninsular Malaysia. Emerg. Infect. Dis. 2001, 7, 439–441. [Google Scholar] [CrossRef]
- Halpin, K.; Hyatt, A.D.; Fogarty, R.; Middleton, D.; Bingham, J.; Epstein, J.H.; Rahman, S.A.; Hughes, T.; Smith, C.; Field, H.E.; et al. Pteropid bats are confirmed as the reservoir hosts of henipaviruses: A comprehensive experimental study of virus transmission. Am. J. Trop. Med. Hyg. 2011, 85, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.T.; Shieh, W.J.; Kumar, S.; Norain, K.; Abdullah, W.; Guarner, J.; Goldsmith, C.S.; Chua, K.B.; Lam, S.K.; Tan, C.T.; et al. Nipah virus infection: Pathology and pathogenesis of an emerging paramyxoviral zoonosis. Am. J. Pathol. 2002, 161, 2153–2167. [Google Scholar] [CrossRef]
- Harit, A.K.; Ichhpujani, R.L.; Gupta, S.; Gill, K.S.; Lal, S.; Ganguly, N.K.; Agarwal, S.P. Nipah/Hendra virus outbreak in Siliguri, West Bengal, India in 2001. Indian J. Med. Res. 2006, 123, 553–560. [Google Scholar] [PubMed]
- Wang, L.-F.; Harcourt, B.H.; Yu, M.; Tamin, A.; Rota, P.A.; Bellini, W.J.; Eaton, B.T. Molecular biology of Hendra and Nipah viruses. Microbes Infect. 2001, 3, 279–287. [Google Scholar] [CrossRef]
- Chua, K.B. Nipah virus outbreak in Malaysia. J. Clin. Virol. 2003, 26, 265–275. [Google Scholar] [CrossRef]
- Marsh, G.A.; de Jong, C.; Barr, J.A.; Tachedjian, M.; Smith, C.; Middleton, D.; Yu, M.; Todd, S.; Foord, A.J.; Haring, V.; et al. Cedar Virus: A Novel Henipavirus Isolated from Australian Bats. PLoS Pathog. 2012, 8, e1002836. [Google Scholar] [CrossRef]
- Ciancanelli, M.J.; Volchkova, V.A.; Shaw, M.L.; Volchkov, V.E.; Basler, C.F. Nipah virus sequesters inactive STAT1 in the nucleus via a P gene-encoded mechanism. J. Virol. 2009, 83, 7828–7841. [Google Scholar] [CrossRef]
- Goh, K.J.; Tan, C.T.; Chew, N.K.; Tan, P.S.; Kamarulzaman, A.; Sarji, S.A.; Wong, K.T.; Abdullah, B.J.; Chua, K.B.; Lam, S.K. Clinical features of Nipah virus encephalitis among pig farmers in Malaysia. N. Engl. J. Med. 2000, 342, 1229–1235. [Google Scholar] [CrossRef]
- Wong, K.T.; Tan, C.T. Clinical and pathological manifestations of human henipavirus infection. Curr. Top. Microbiol. Immunol. 2012, 359, 95–104. [Google Scholar]
- Maisner, A.; Neufeld, J.; Weingartl, H. Organ- and endotheliotropism of Nipah virus infections in vivo and in vitro. Thromb. Haemost. 2009, 102, 1014–1023. [Google Scholar]
- Pernet, O.; Wang, Y.E.; Lee, B. Henipavirus receptor usage and tropism. Curr. Top. Microbiol. Immunol. 2012, 359, 59–78. [Google Scholar]
- Lieu, K.G.; Marsh, G.A.; Wang, L.F.; Netter, H.J. The non-pathogenic Henipavirus Cedar paramyxovirus phosphoprotein has a compromised ability to target STAT1 and STAT2. Antivir. Res. 2015, 124, 69–76. [Google Scholar] [CrossRef]
- Wong, K.T.; Grosjean, I.; Brisson, C.; Blanquier, B.; Fevre-Montange, M.; Bernard, A.; Loth, P.; Georges-Courbot, M.-C.C.; Chevallier, M.; Akaoka, H.; et al. A golden hamster model for human acute Nipah virus infection. Am. J. Pathol. 2003, 163, 2127–2137. [Google Scholar] [CrossRef]
- Rockx, B.; Brining, D.; Kramer, J.; Callison, J.; Ebihara, H.; Mansfield, K.; Feldmann, H. Clinical Outcome of Henipavirus Infection in Hamsters is Determined by the Route and Dose of Infection. J. Virol. 2011, 85, 7658–7671. [Google Scholar] [CrossRef]
- Harcourt, B.H.; Lowe, L.; Tamin, A.; Liu, X.; Bankamp, B.; Bowden, N.; Rollin, P.E.; Comer, J.A.; Ksiazek, T.G.; Hossain, M.J.; et al. Genetic characterization of Nipah virus, Bangladesh, 2004. Emerg. Infect. Dis. 2005, 11, 1594–1597. [Google Scholar] [CrossRef]
- McGuire, A.; Miedema, K.; Fauver, J.R.; Rico, A.; Aboellail, T.; Quackenbush, S.L.; Hawkinson, A.; Schountz, T. Maporal hantavirus causes mild pathology in deer mice (Peromyscus maniculatus). Viruses 2016, 8, 286. [Google Scholar] [CrossRef]
- Schountz, T.; Shaw, T.I.; Glenn, T.C.; Feldmann, H.; Prescott, J. Expression profiling of lymph node cells from deer mice infected with Andes virus. BMC Immunol. 2013, 14, 18. [Google Scholar] [CrossRef]
- Stanwick, T.L.; Hallum, J.V. Role of interferon in six cell lines persistently infected with rubella virus. Infect. Immun. 1974, 10, 810–815. [Google Scholar]
- DeBuysscher, B.L.; Scott, D.; Thomas, T.; Feldmann, H.; Prescott, J. Peri-exposure protection against Nipah virus disease using a single-dose recombinant vesicular stomatitis virus-based vaccine. Npj Vaccines 2016, 1, 16002. [Google Scholar] [CrossRef]
- Shaw, M.L.; Cardenas, W.B.; Zamarin, D.; Palese, P.; Basler, C.F. Nuclear localization of the Nipah virus W protein allows for inhibition of both virus- and toll-like receptor 3-triggered signaling pathways. J. Virol. 2005, 79, 6078–6088. [Google Scholar] [CrossRef]
- Harcourt, B.H.; Tamin, A.; Ksiazek, T.G.; Rollin, P.E.; Anderson, L.J.; Bellini, W.J.; Rota, P.A. Molecular characterization of Nipah virus, a newly emergent paramyxovirus. Virology 2000, 271, 334–349. [Google Scholar] [CrossRef]
- Yoneda, M.; Guillaume, V.; Sato, H.; Fujita, K.; Georges-Courbot, M.C.; Ikeda, F.; Omi, M.; Muto-Terao, Y.; Wild, T.F.; Kai, C. The nonstructural proteins of Nipah virus play a key role in pathogenicity in experimentally infected animals. PLoS ONE 2010, 5, e12709. [Google Scholar] [CrossRef]
- Shaw, M.L.; Garcia-Sastre, A.; Palese, P.; Basler, C.F. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J. Virol. 2004, 78, 5633–5641. [Google Scholar] [CrossRef]
- Andrejeva, J.; Childs, K.S.; Young, D.F.; Carlos, T.S.; Stock, N.; Goodbourn, S.; Randall, R.E. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. USA 2004, 101, 17264–17269. [Google Scholar] [CrossRef]
- Nagai, Y.; Ito, Y.; Hamaguchi, M.; Yoshida, T.; Matsumoto, T. Relation of interferon production to the limited replication of Newcastle disease virus in L cells. J. Gen. Virol. 1981, 55, 109–116. [Google Scholar] [CrossRef]
- Folimonova, S.Y. Superinfection Exclusion Is an Active Virus-Controlled Function That Requires a Specific Viral Protein. J. Virol. 2012, 86, 5554–5561. [Google Scholar] [CrossRef]
- Negrete, O.A.; Levroney, E.L.; Aguilar, H.C.; Bertolotti-Ciarlet, A.; Nazarian, R.; Tajyar, S.; Lee, B. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 2005, 436, 401–405. [Google Scholar] [CrossRef]
- Laing, E.D.; Amaya, M.; Navaratnarajah, C.K.; Feng, Y.R.; Cattaneo, R.; Wang, L.F.; Broder, C.C. Rescue and characterization of recombinant cedar virus, a non-pathogenic Henipavirus species. Virol. J. 2018, 15, 56. [Google Scholar] [CrossRef]
- Burroughs, A.L.; Durr, P.A.; Boyd, V.; Graham, K.; White, J.R.; Todd, S.; Barr, J.; Smith, I.; Baverstock, G.; Meers, J.; et al. Hendra virus infection dynamics in the grey-headed flying fox (Pteropus poliocephalus) at the southern-most extent of its range: Further evidence this species does not readily transmit the virus to horses. PLoS ONE 2016, 11, e0155252. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schountz, T.; Campbell, C.; Wagner, K.; Rovnak, J.; Martellaro, C.; DeBuysscher, B.L.; Feldmann, H.; Prescott, J. Differential Innate Immune Responses Elicited by Nipah Virus and Cedar Virus Correlate with Disparate In Vivo Pathogenesis in Hamsters. Viruses 2019, 11, 291. https://doi.org/10.3390/v11030291
Schountz T, Campbell C, Wagner K, Rovnak J, Martellaro C, DeBuysscher BL, Feldmann H, Prescott J. Differential Innate Immune Responses Elicited by Nipah Virus and Cedar Virus Correlate with Disparate In Vivo Pathogenesis in Hamsters. Viruses. 2019; 11(3):291. https://doi.org/10.3390/v11030291
Chicago/Turabian StyleSchountz, Tony, Corey Campbell, Kaitlyn Wagner, Joel Rovnak, Cynthia Martellaro, Blair L DeBuysscher, Heinz Feldmann, and Joseph Prescott. 2019. "Differential Innate Immune Responses Elicited by Nipah Virus and Cedar Virus Correlate with Disparate In Vivo Pathogenesis in Hamsters" Viruses 11, no. 3: 291. https://doi.org/10.3390/v11030291
APA StyleSchountz, T., Campbell, C., Wagner, K., Rovnak, J., Martellaro, C., DeBuysscher, B. L., Feldmann, H., & Prescott, J. (2019). Differential Innate Immune Responses Elicited by Nipah Virus and Cedar Virus Correlate with Disparate In Vivo Pathogenesis in Hamsters. Viruses, 11(3), 291. https://doi.org/10.3390/v11030291