Initial Characterization of the Epstein–Barr Virus BSRF1 Gene Product


 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Construction of Expression Plasmids
2.3. Construction of BSRF1-KO EBV-BAC and Cloning of HEK293 Cells Maintaining EBV-BAC
2.4. Transfection, Immunoblotting, Viral DNA Quantification, and Progeny Titration
2.5. Transformation Assay
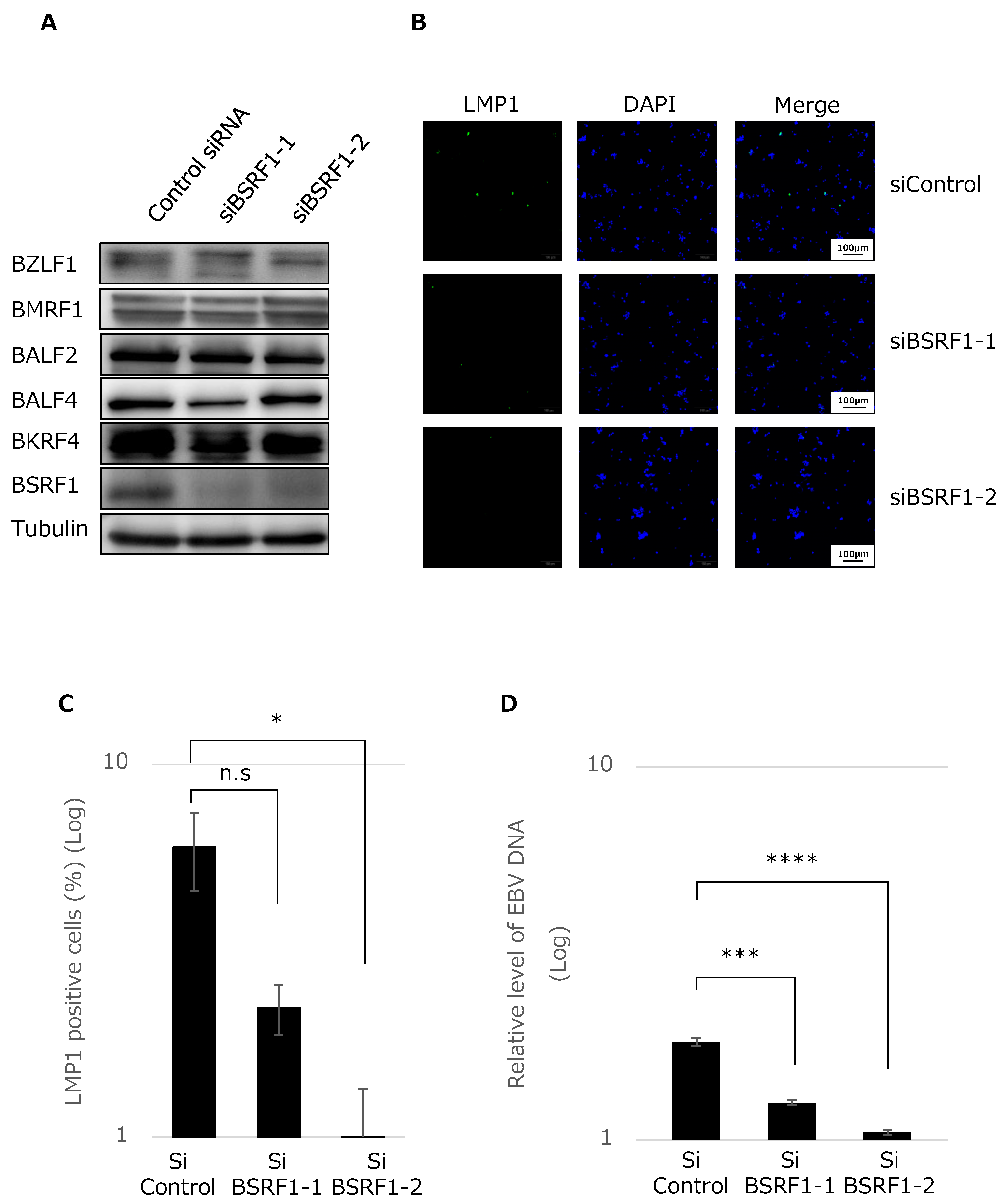
2.6. Knockdown of BSRF1 by siRNA
2.7. Immunofluorescence Analysis
2.8. Immunoprecipitation
2.9. Fractionation of Extracellular Virions
3. Results
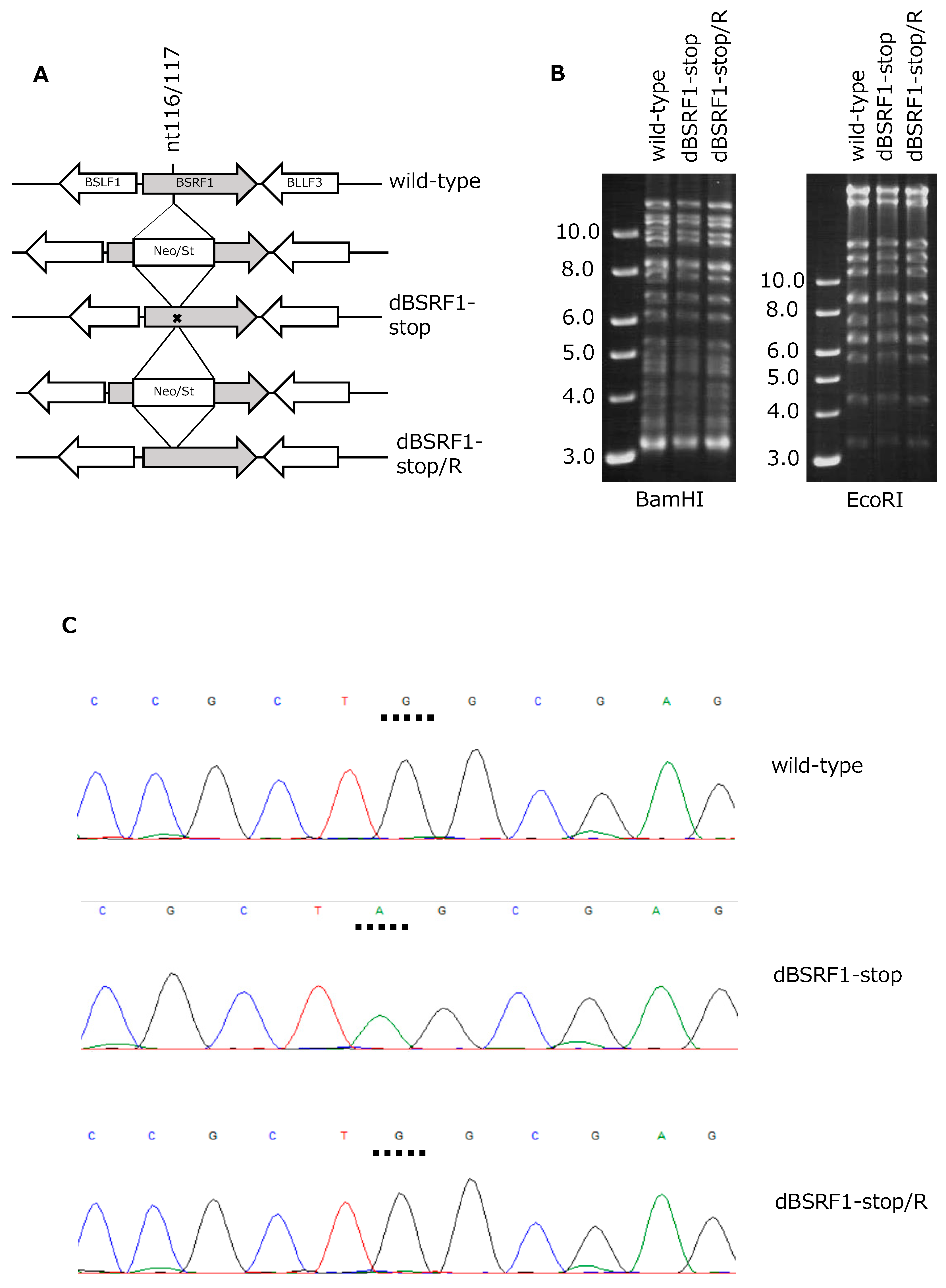
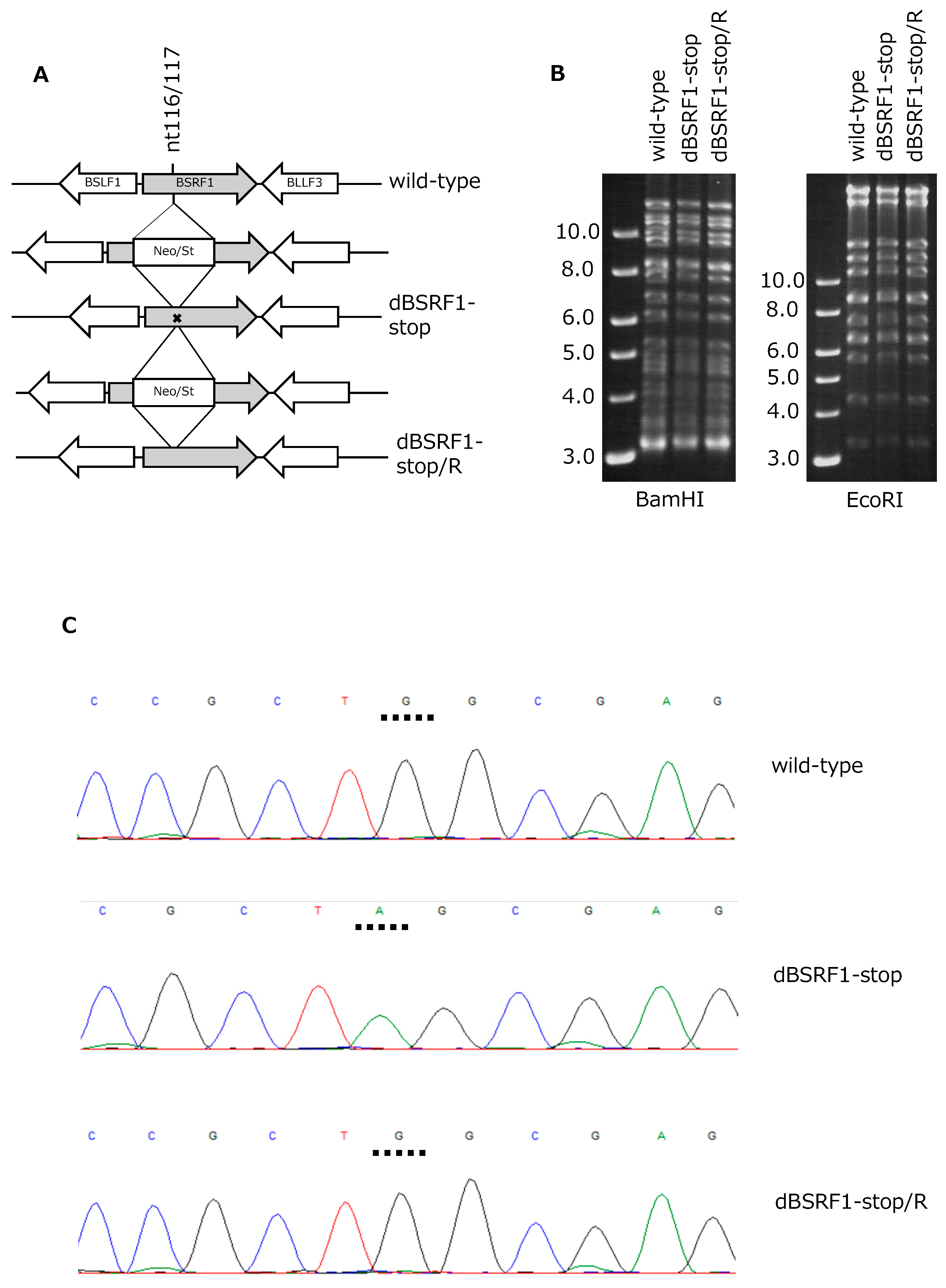
3.1. Preparation of BSRF1-Knockout EBV
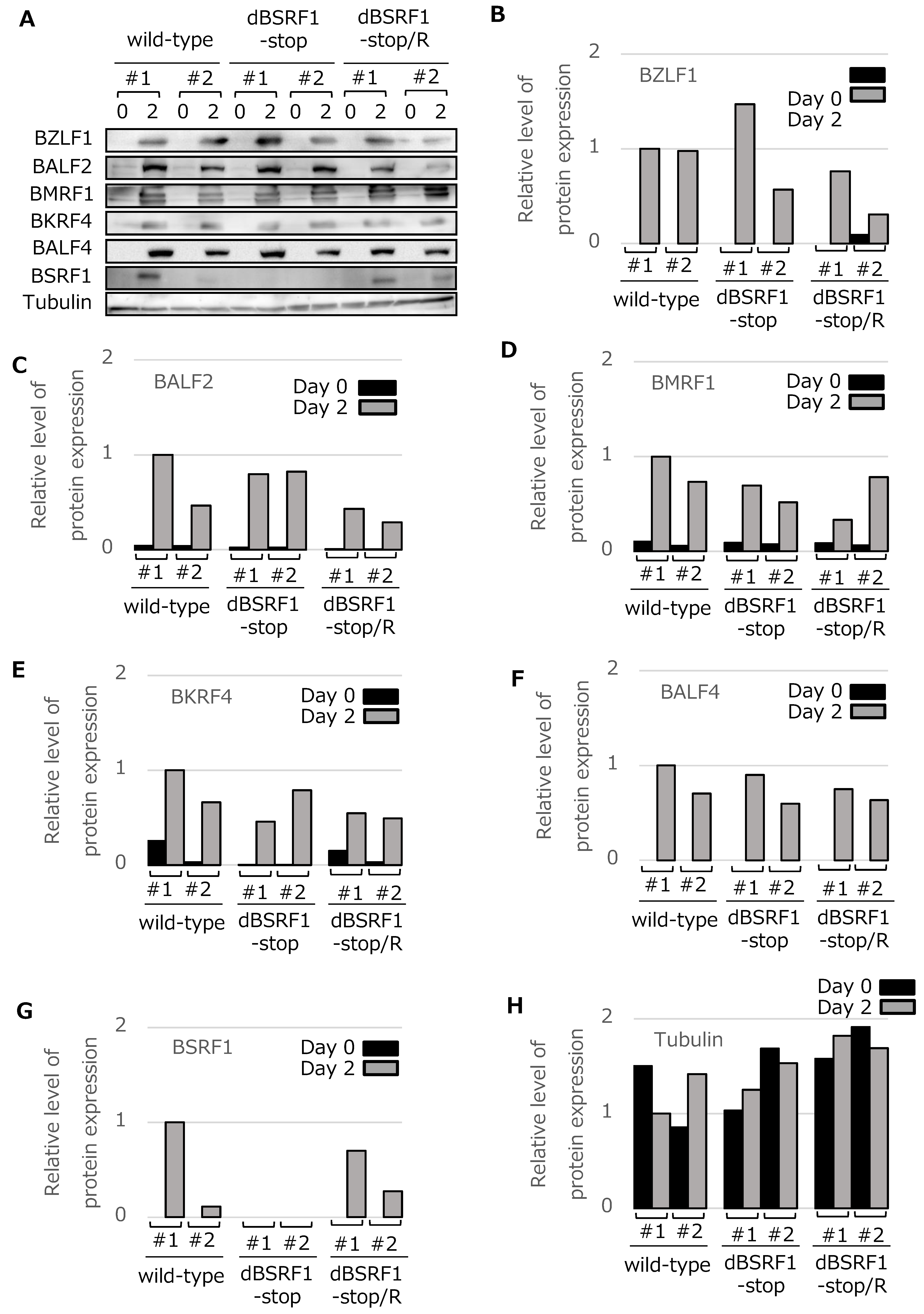
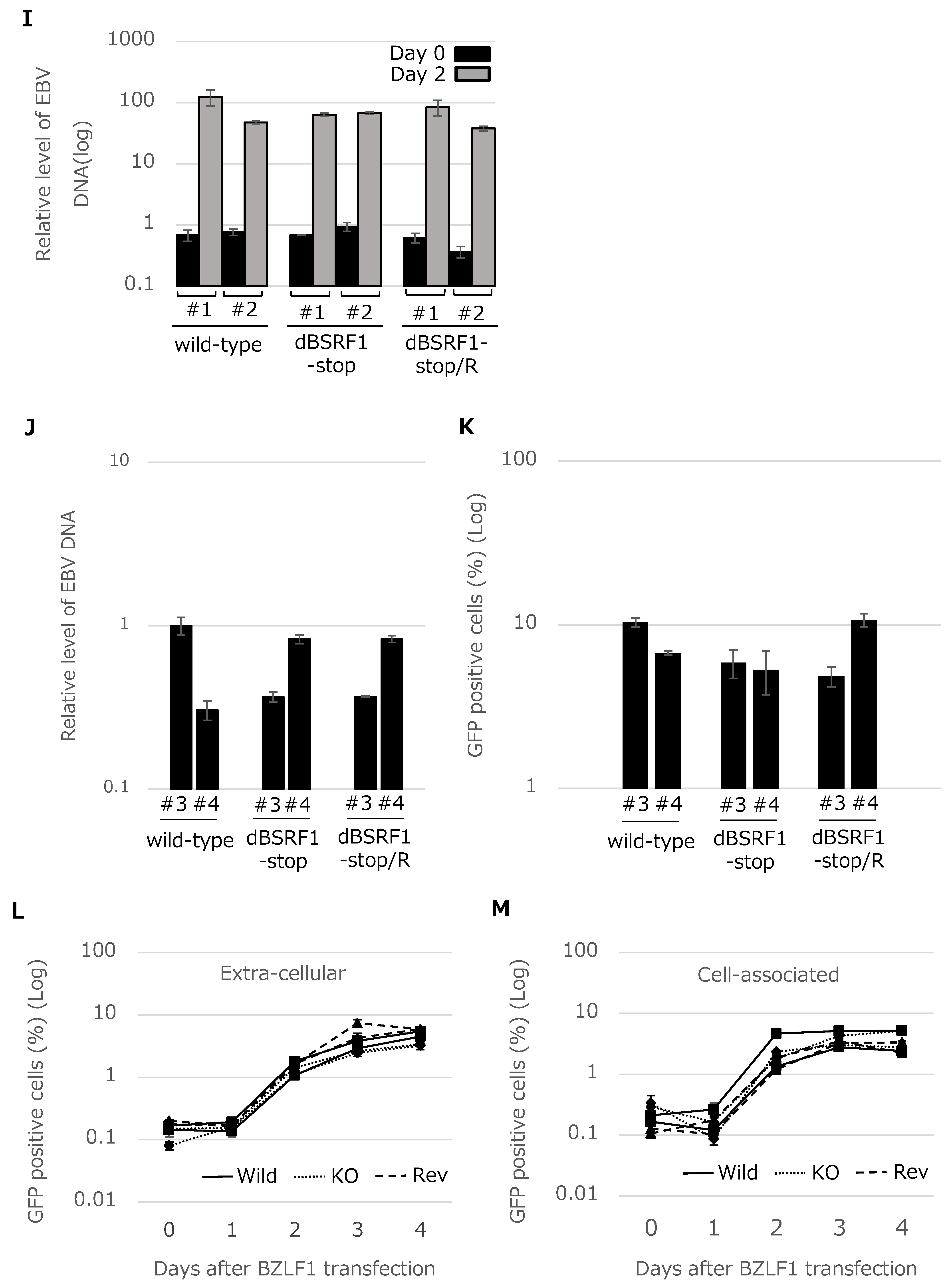
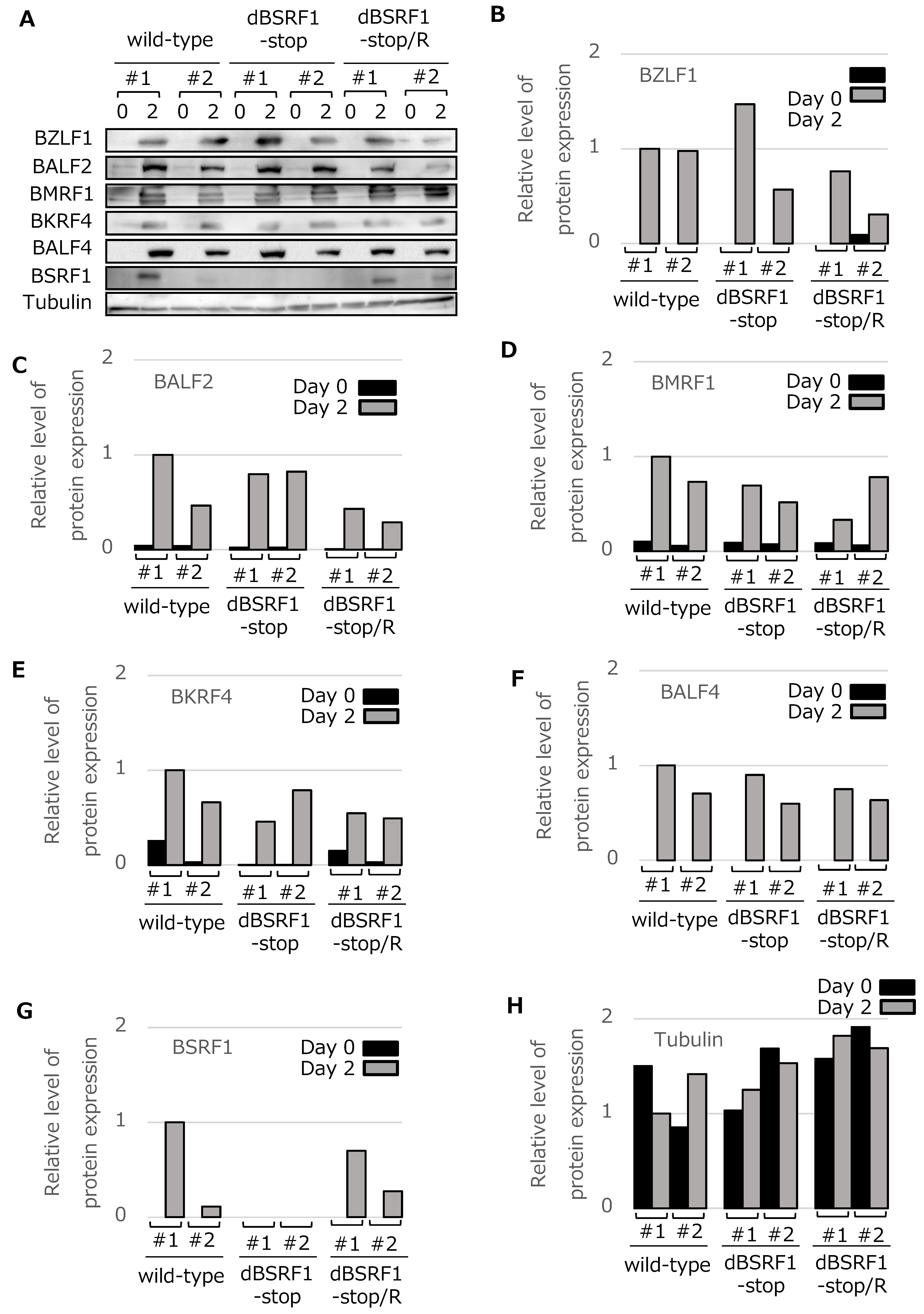
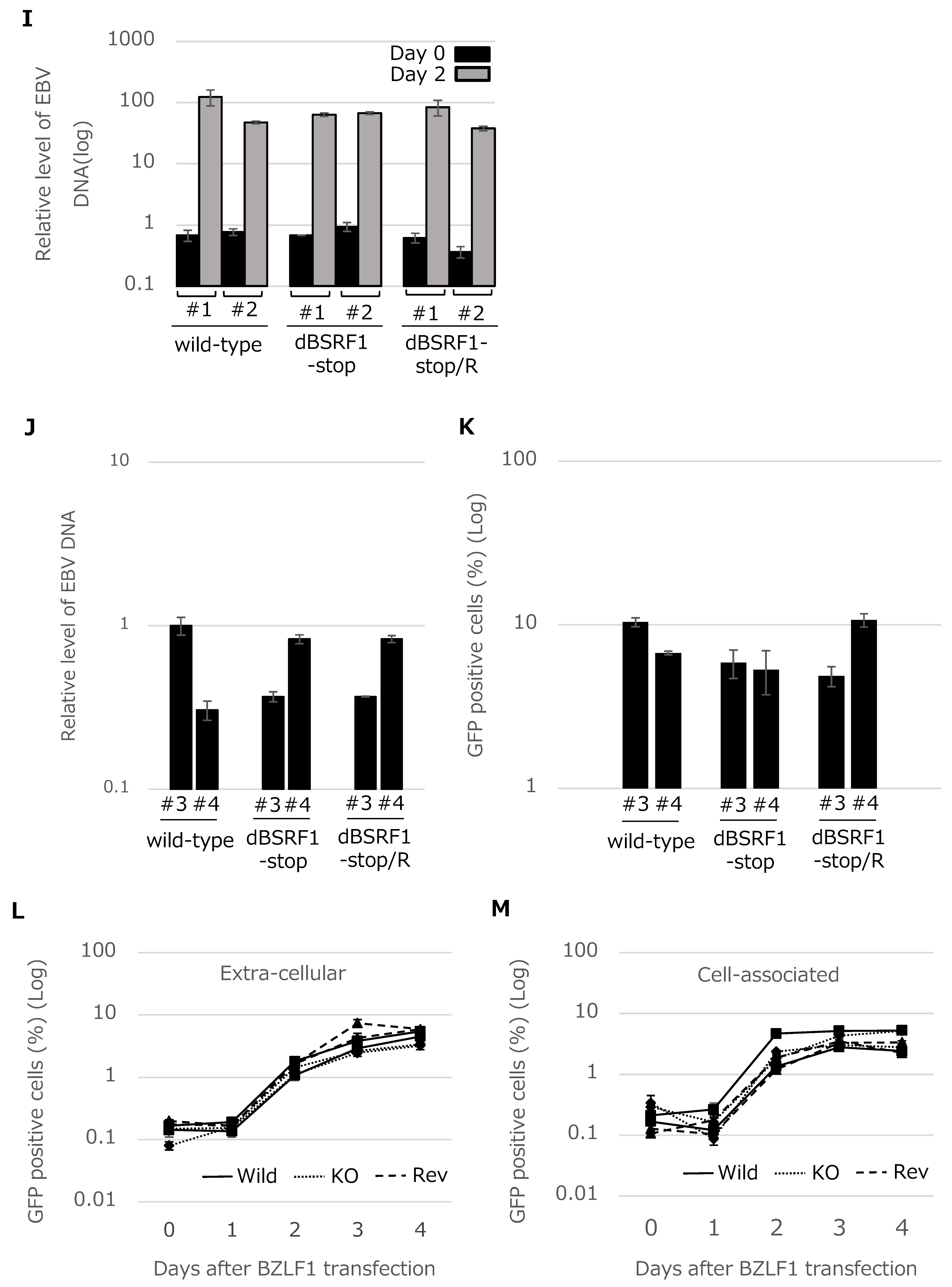
3.2. BSRF1 Is Not Required for EBV Replication in HEK293 Cells
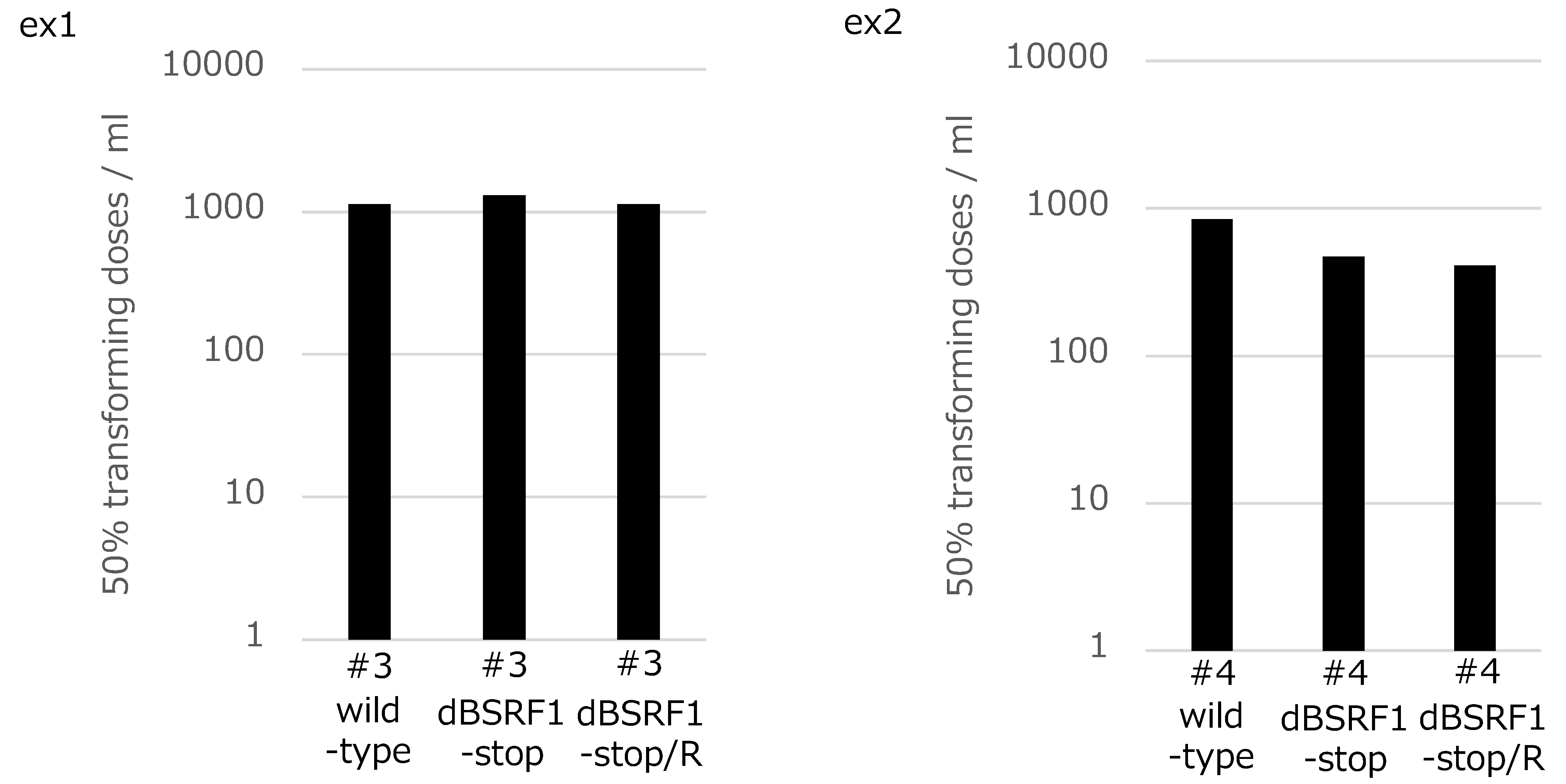
3.3. BSRF1 Is Not Required for B-Cell Transformation
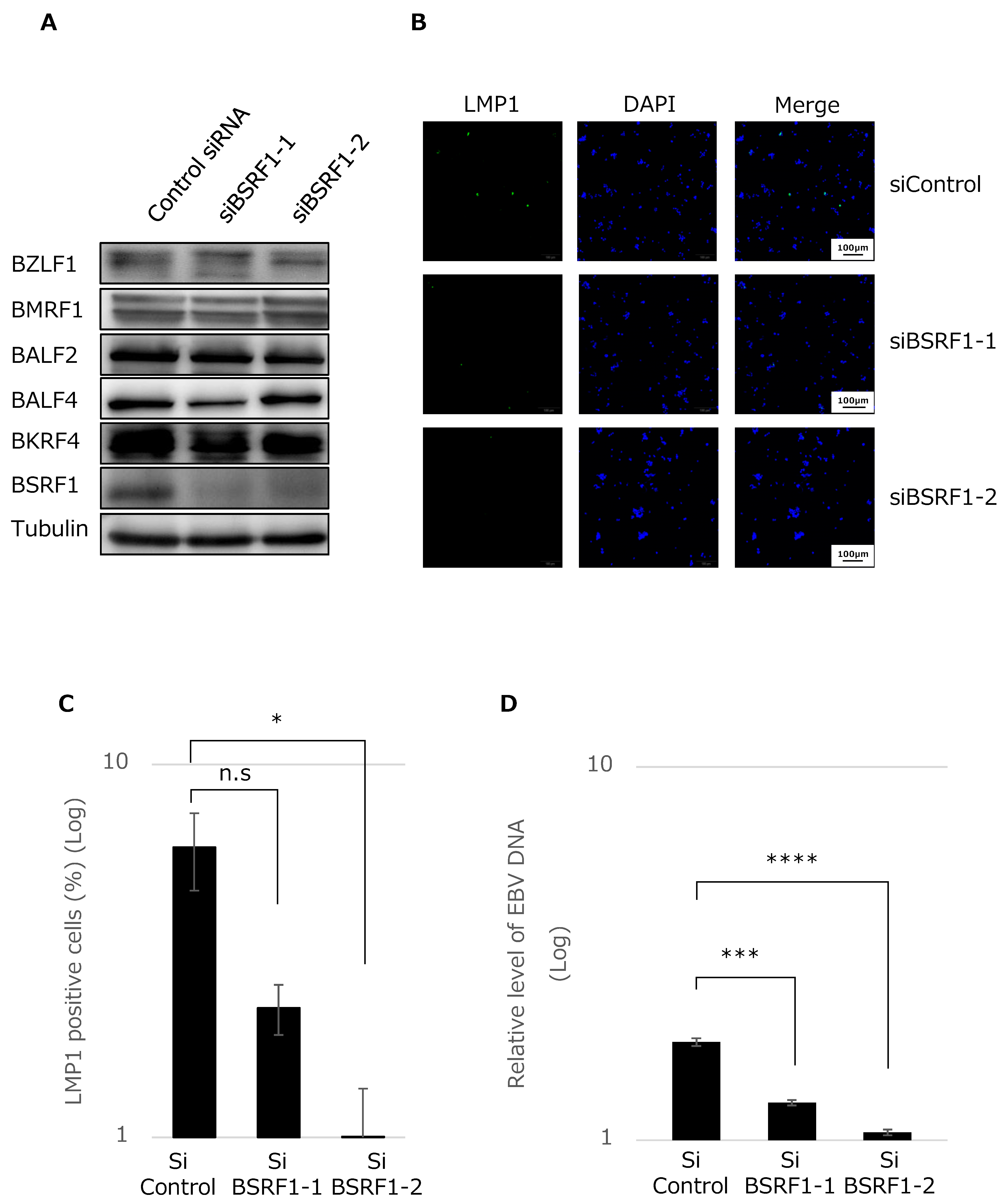
3.4. BSRF1 Is Required for Efficient Progeny Production in B95-8 Cells
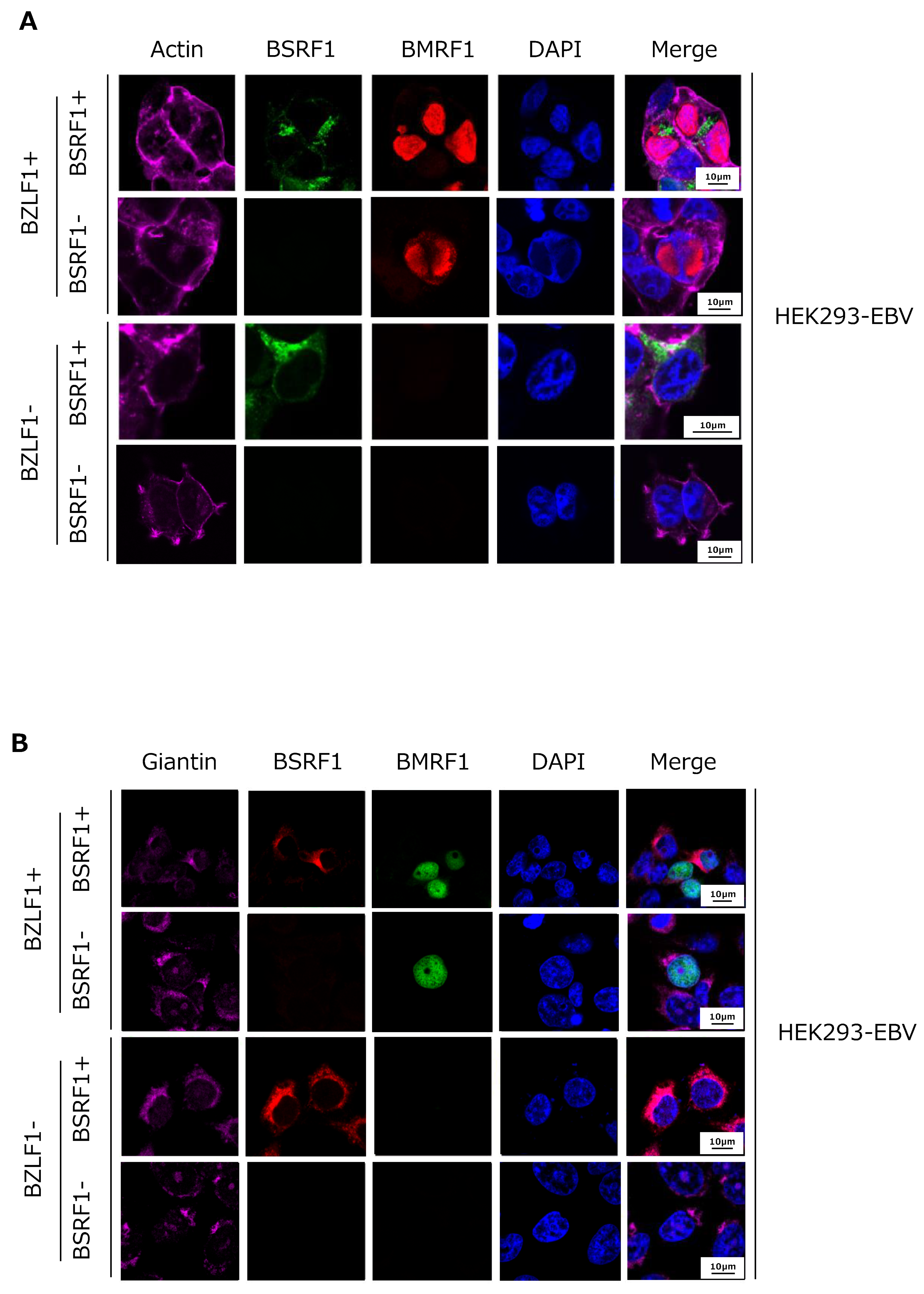
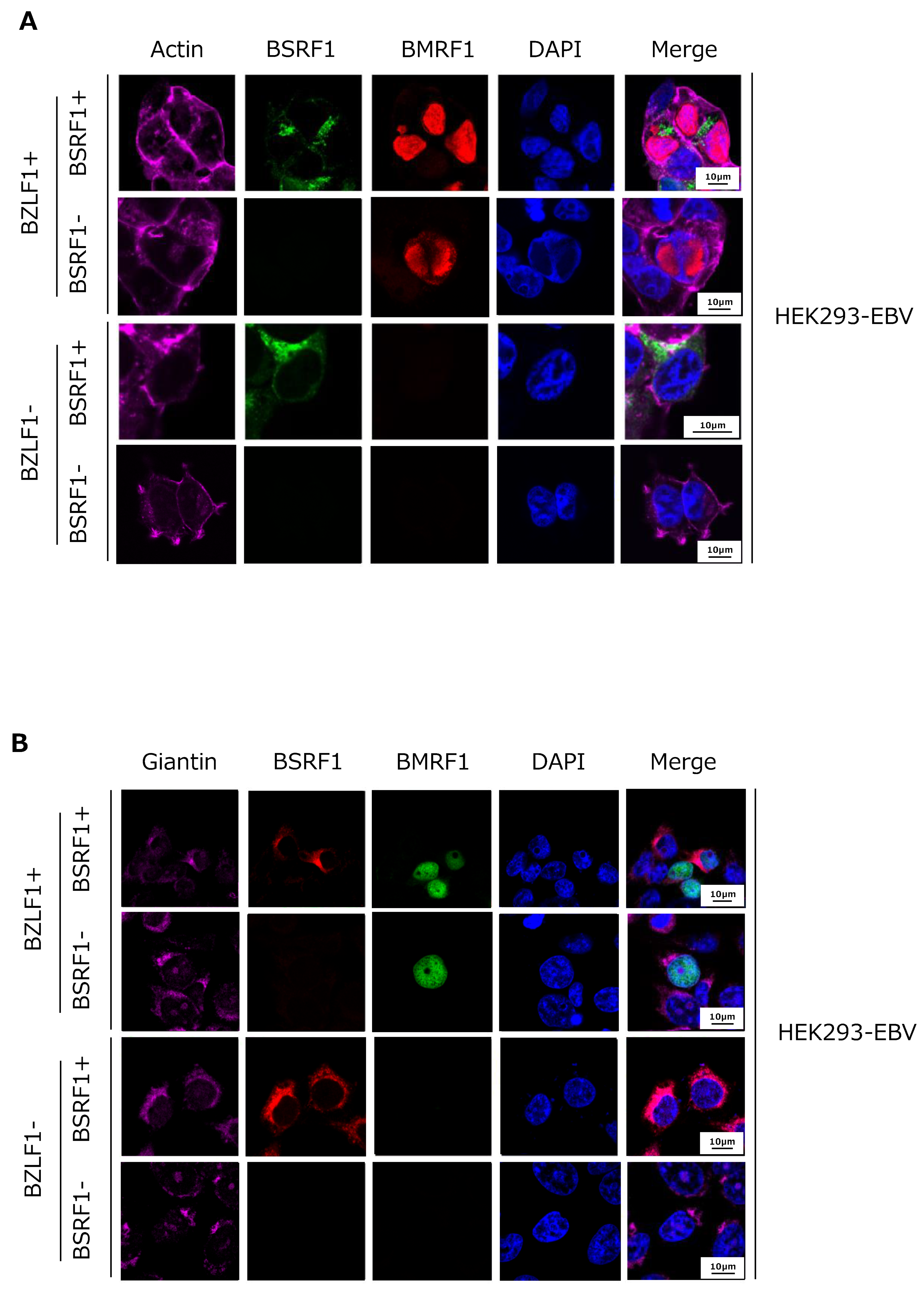
3.5. Cytoplasmic Localization of BSRF1 Protein
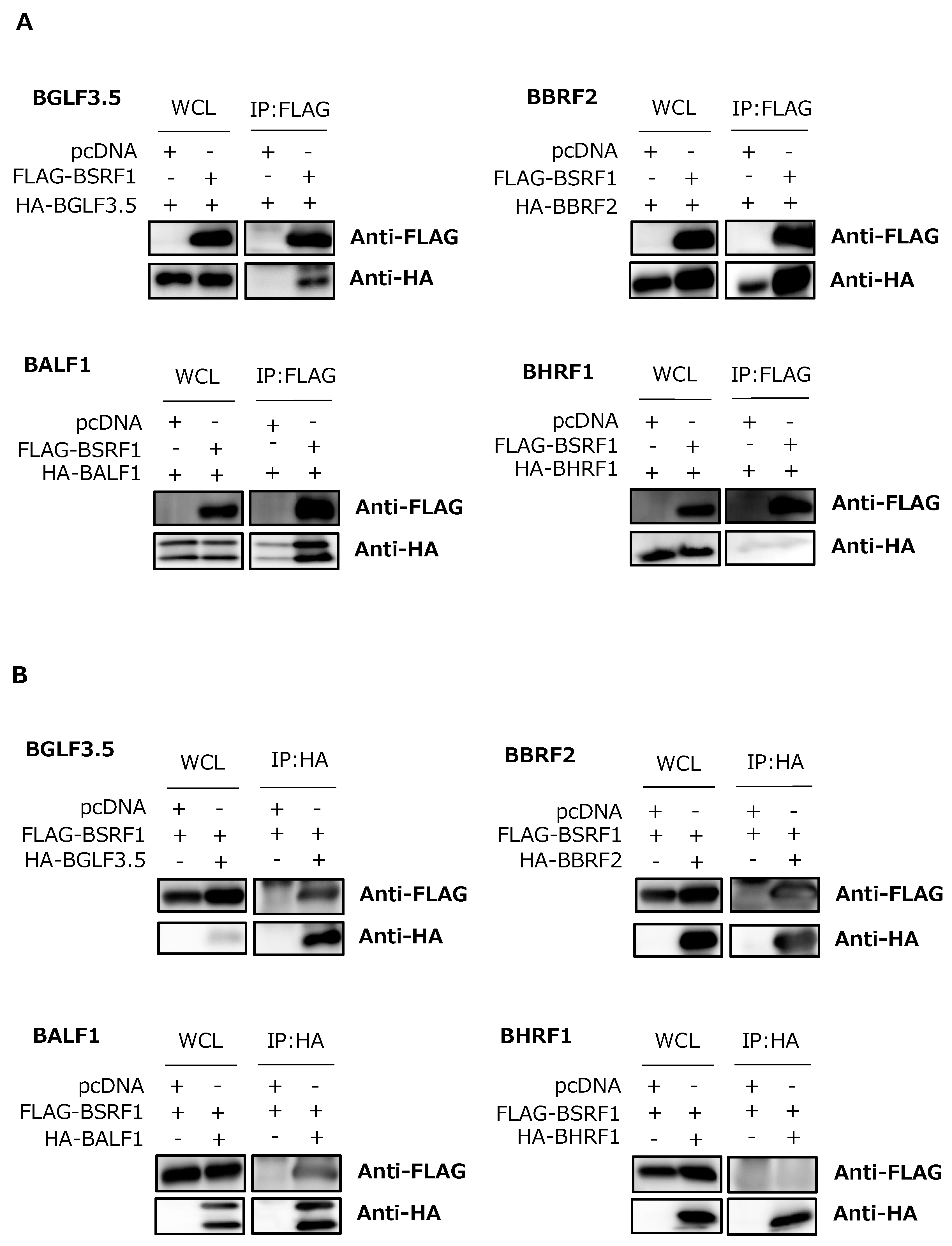
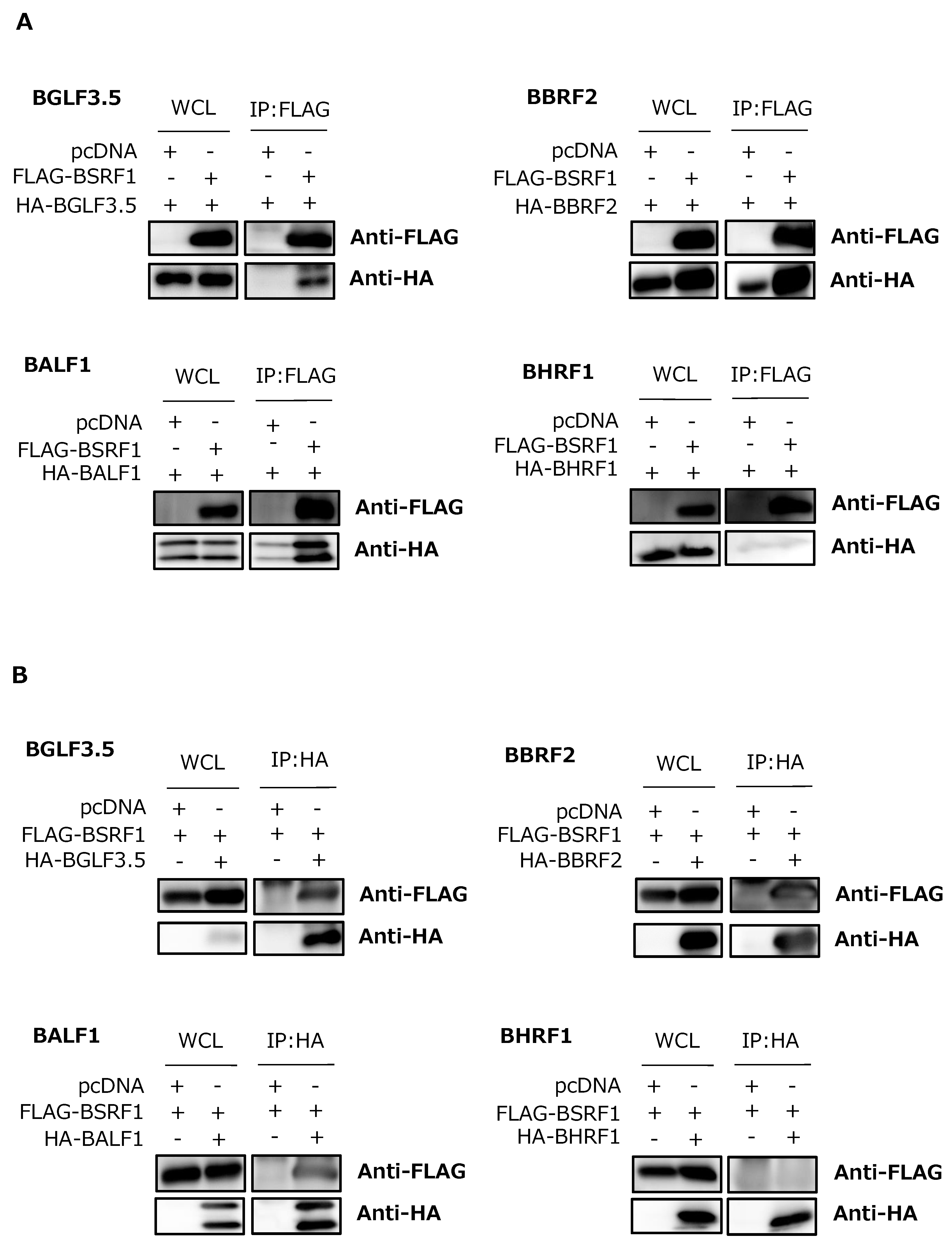
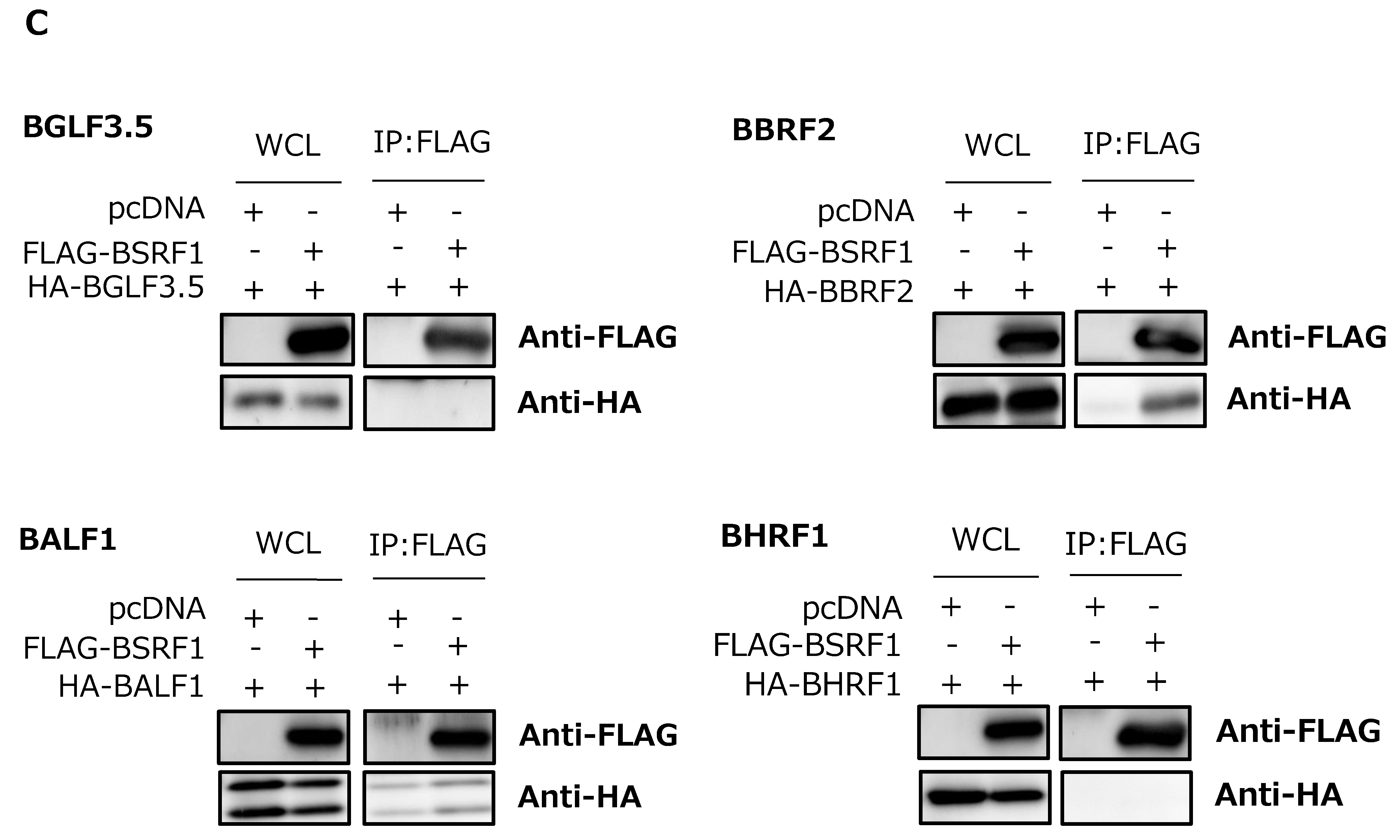
3.6. Association of BSRF1 with Other Viral Proteins
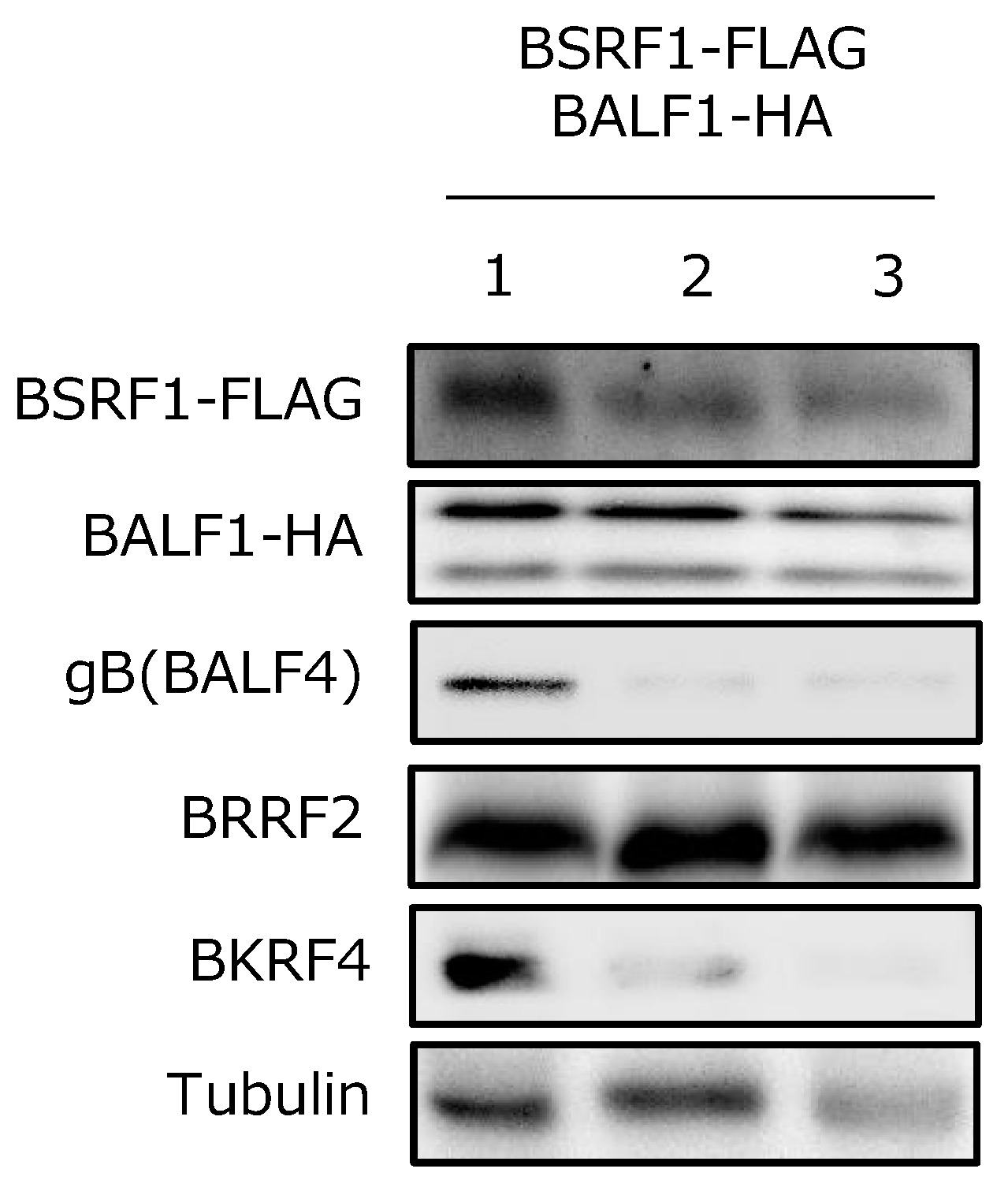
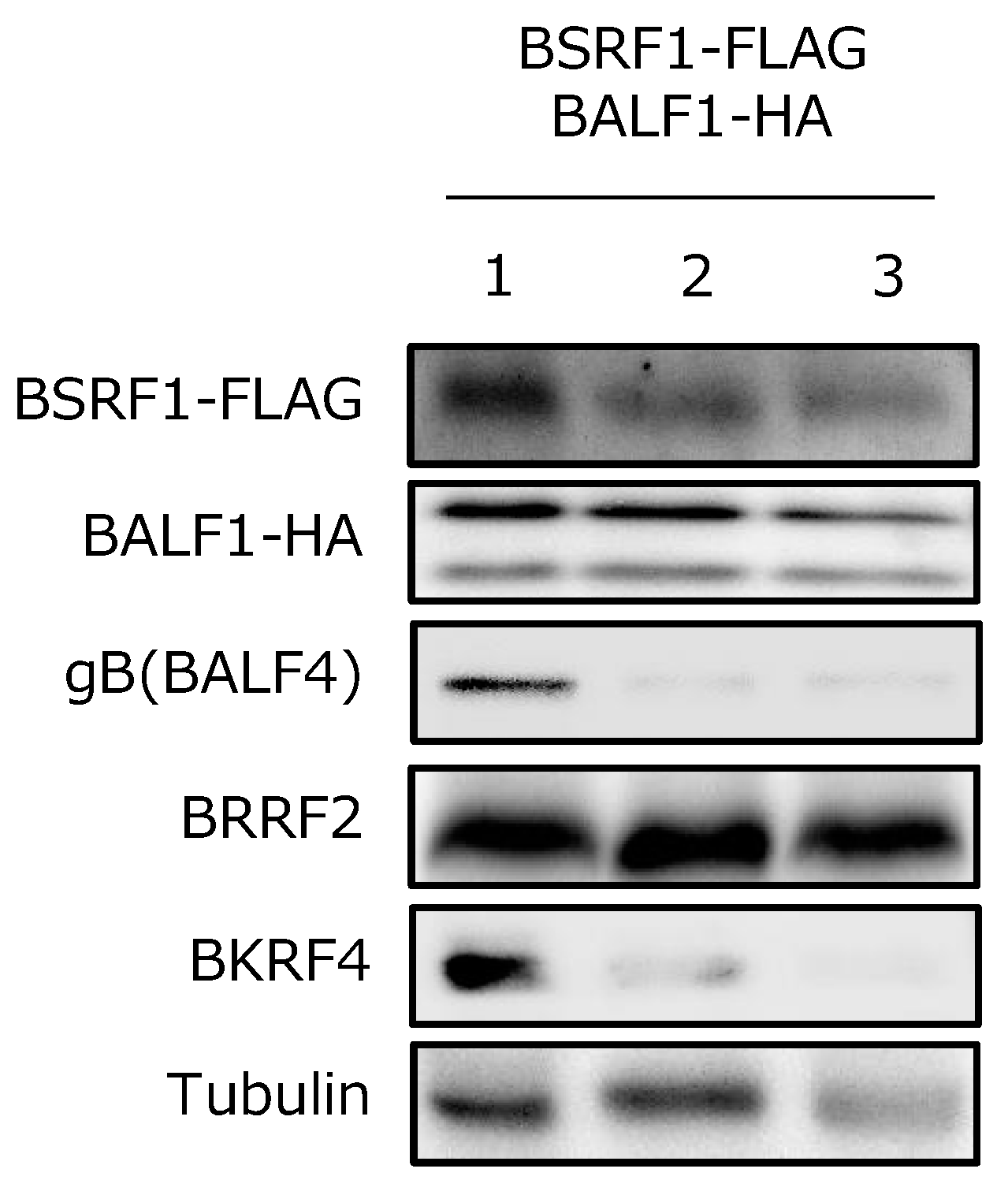
3.7. BSRF1 Was Present in Virions with BALF1
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Dunmire, S.K.; Hogquist, K.A.; Balfour, H.H. Infectious mononucleosis. Curr. Top. Microbiol. Immunol. 2015, 390, 211–240. [Google Scholar] [PubMed]
- Shah, K.M.; Young, L.S. Epstein–Barr virus and carcinogenesis: Beyond Burkitt’s lymphoma. Clin. Microbiol. Infect. 2009, 15, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Sato, Y.; Kimura, H. Modes of infection and oncogenesis by the Epstein–Barr virus. Rev. Med. Virol. 2014, 24, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med. Virol. 2014, 24, 142–153. [Google Scholar] [CrossRef]
- Murata, T. Regulation of Epstein–Barr virus reactivation from latency. Microbiol. Immunol. 2014, 58, 307–317. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, J.; El-Guindy, A. Epstein–Barr virus lytic cycle reactivation. Curr. Top. Microbiol. Immunol. 2015, 391, 237–261. [Google Scholar]
- Murata, T. Encyclopedia of EBV-encoded lytic genes: An update. Adv. Exp. Med. Biol. 2018, 1045, 395–412. [Google Scholar] [PubMed]
- Sathish, N.; Wang, X.; Yuan, Y. Tegument proteins of Kaposi’s sarcoma-associated herpesvirus and related gamma-herpesviruses. Front. Microbiol. 2012, 3, 98. [Google Scholar] [CrossRef]
- Johannsen, E.; Luftig, M.; Chase, M.R.; Weicksel, S.; Cahir-McFarland, E.; Illanes, D.; Sarracino, D.; Kieff, E. Proteins of purified Epstein–Barr virus. Proc. Natl. Acad. Sci. USA 2004, 101, 16286–16291. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Oda, S.; Watanabe, M.; Oyama, M.; Kozuka-Hata, H.; Koyanagi, N.; Maruzuru, Y.; Arii, J.; Kawaguchi, Y. Roles of the phosphorylation of herpes simplex virus 1 UL51 at a specific site in viral replication and pathogenicity. J. Virol. 2018, 92, e01035-18. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, N.; Daikoku, T.; Koshizuka, T.; Yamauchi, Y.; Yoshikawa, T.; Nishiyama, Y. Subcellular localization of herpes simplex virus type 1 UL51 protein and role of palmitoylation in Golgi apparatus targeting. J. Virol. 2003, 77, 3204–3216. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, N.; Kawaguchi, Y.; Tanaka, M.; Kato, A.; Kato, A.; Kimura, H.; Nishiyama, Y. Herpes simplex virus type 1 UL51 protein is involved in maturation and egress of virus particles. J. Virol. 2005, 79, 6947–6956. [Google Scholar] [CrossRef] [PubMed]
- Roller, R.J.; Fetters, R. The herpes simplex virus 1 UL51 protein interacts with the UL7 protein and plays a role in its recruitment into the virion. J. Virol. 2015, 89, 3112–3122. [Google Scholar] [CrossRef] [PubMed]
- Dietz, A.N.; Villinger, C.; Becker, S.; Frick, M.; von Einem, J. A tyrosine-based trafficking motif of the tegument protein pUL71 is crucial for human cytomegalovirus secondary envelopment. J. Virol. 2018, 92, e00907-17. [Google Scholar] [CrossRef] [PubMed]
- Schauflinger, M.; Fischer, D.; Schreiber, A.; Chevillotte, M.; Walther, P.; Mertens, T.; von Einem, J. The tegument protein UL71 of human cytomegalovirus is involved in late envelopment and affects multivesicular bodies. J. Virol. 2011, 85, 3821–3832. [Google Scholar] [CrossRef] [PubMed]
- Masud, H.; Watanabe, T.; Yoshida, M.; Sato, Y.; Goshima, F.; Kimura, H.; Murata, T. Epstein–Barr virus BKRF4 gene product is required for efficient progeny production. J. Virol. 2017, 91, e00975-17. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Isomura, H.; Yamashita, Y.; Toyama, S.; Sato, Y.; Nakayama, S.; Kudoh, A.; Iwahori, S.; Kanda, T.; Tsurumi, T. Efficient production of infectious viruses requires enzymatic activity of Epstein–Barr virus protein kinase. Virology 2009, 389, 75–81. [Google Scholar] [CrossRef]
- Konishi, N.; Narita, Y.; Hijioka, F.; Masud, H.; Sato, Y.; Kimura, H.; Murata, T. BGLF2 Increases infectivity of Epstein–Barr virus by activating AP-1 upon de novo infection. mSphere 2018, 3, e00138-18. [Google Scholar] [CrossRef]
- Delecluse, H.J.; Hilsendegen, T.; Pich, D.; Zeidler, R.; Hammerschmidt, W. Propagation and recovery of intact, infectious Epstein–Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. USA 1998, 95, 8245–8250. [Google Scholar] [CrossRef]
- Narita, Y.; Murata, T.; Ryo, A.; Kawashima, D.; Sugimoto, A.; Kanda, T.; Kimura, H.; Tsurumi, T. Pin1 interacts with the Epstein–Barr virus DNA polymerase catalytic subunit and regulates viral DNA replication. J. Virol. 2013, 87, 2120–2127. [Google Scholar] [CrossRef]
- Linstedt, A.D.; Hauri, H.P. Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol. Biol. Cell 1993, 4, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Arii, J.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. The interaction between herpes simplex virus 1 tegument proteins UL51 and UL14 and its role in virion morphogenesis. J. Virol. 2016, 90, 8754–8767. [Google Scholar] [CrossRef]
- Liang, Q.; Wei, D.; Chung, B.; Brulois, K.F.; Guo, C.; Dong, S.; Gao, S.J.; Feng, P.; Liang, C.; Jung, J.U. Novel role of vBcl2 in the virion assembly of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2018, 92, e00914-17. [Google Scholar] [CrossRef] [PubMed]
- Song, M.J.; Hwang, S.; Wong, W.H.; Wu, T.T.; Lee, S.; Liao, H.I.; Sun, R. Identification of viral genes essential for replication of murine gamma-herpesvirus 68 using signature-tagged mutagenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 3805–3810. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Fuse, K.; Takano, T.; Narita, Y.; Goshima, F.; Kimura, H.; Murata, T. Roles of Epstein–Barr virus BGLF3.5 gene and two upstream open reading frames in lytic viral replication in HEK293 cells. Virology 2015, 483, 44–53. [Google Scholar] [CrossRef]
- Yoshida, M.; Watanabe, T.; Narita, Y.; Sato, Y.; Goshima, F.; Kimura, H.; Murata, T. The Epstein–Barr virus BRRF1 gene is dispensable for viral replication in HEK293 cells and transformation. Sci. Rep. 2017, 7, 6044. [Google Scholar] [CrossRef]
- Marshall, W.L.; Yim, C.; Gustafson, E.; Graf, T.; Sage, D.R.; Hanify, K.; Williams, L.; Fingeroth, J.; Finberg, R.W. Epstein–Barr virus encodes a novel homolog of the bcl-2 oncogene that inhibits apoptosis and associates with Bax and Bak. J. Virol. 1999, 73, 5181–5185. [Google Scholar]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanagi, Y.; Masud, H.M.A.A.; Watanabe, T.; Sato, Y.; Goshima, F.; Kimura, H.; Murata, T. Initial Characterization of the Epstein–Barr Virus BSRF1 Gene Product. Viruses 2019, 11, 285. https://doi.org/10.3390/v11030285
Yanagi Y, Masud HMAA, Watanabe T, Sato Y, Goshima F, Kimura H, Murata T. Initial Characterization of the Epstein–Barr Virus BSRF1 Gene Product. Viruses. 2019; 11(3):285. https://doi.org/10.3390/v11030285
Chicago/Turabian StyleYanagi, Yusuke, H. M. Abdullah Al Masud, Takahiro Watanabe, Yoshitaka Sato, Fumi Goshima, Hiroshi Kimura, and Takayuki Murata. 2019. "Initial Characterization of the Epstein–Barr Virus BSRF1 Gene Product" Viruses 11, no. 3: 285. https://doi.org/10.3390/v11030285
APA StyleYanagi, Y., Masud, H. M. A. A., Watanabe, T., Sato, Y., Goshima, F., Kimura, H., & Murata, T. (2019). Initial Characterization of the Epstein–Barr Virus BSRF1 Gene Product. Viruses, 11(3), 285. https://doi.org/10.3390/v11030285