Metagenomic Analysis Reveals Three Novel and Prevalent Mosquito Viruses from a Single Pool of Aedes vexans nipponii Collected in the Republic of Korea

 ,
,
Abstract
1. Introduction
2. Materials and Methods
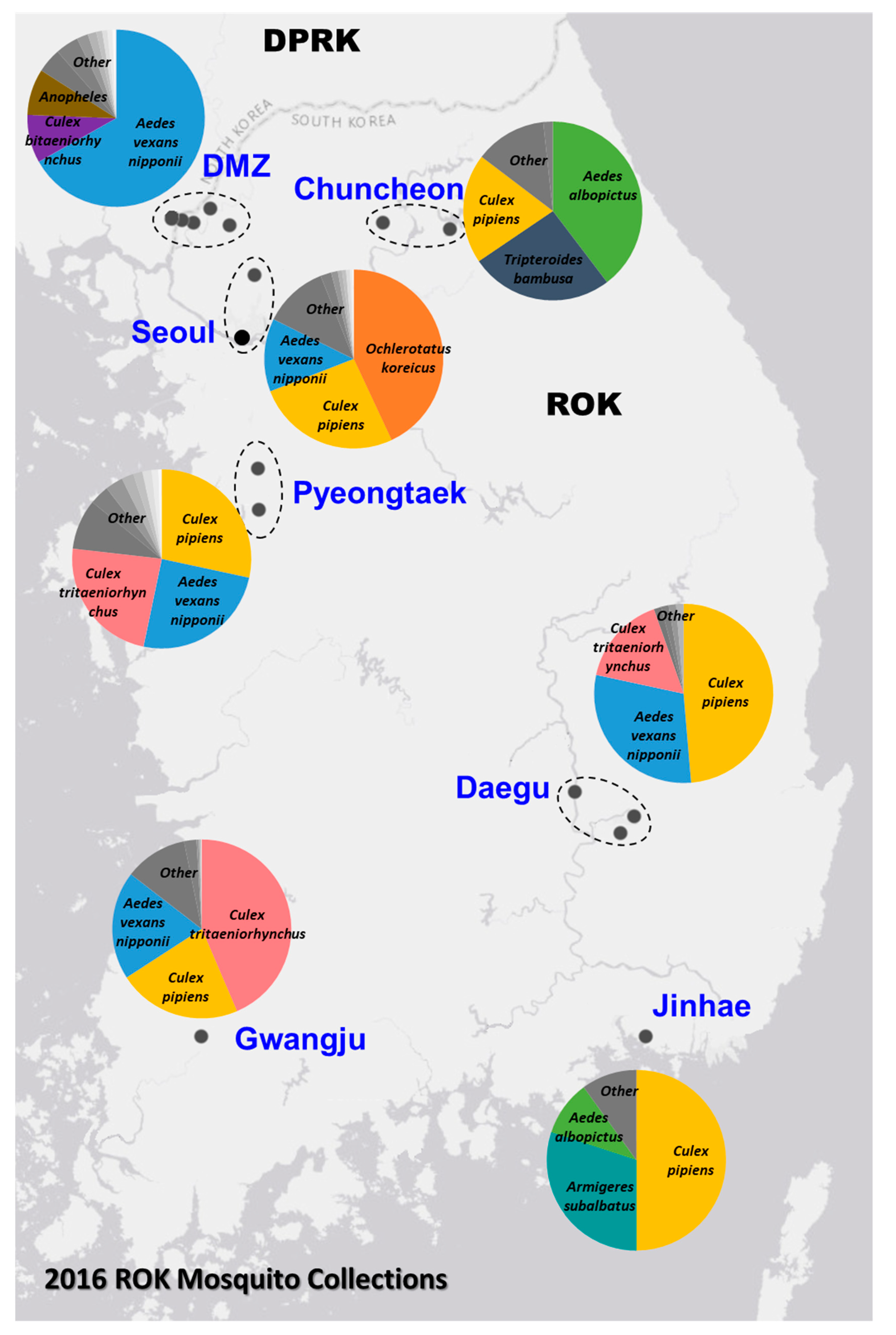
2.1. Mosquito Collections
2.2. Nucleic Acid Extraction, Random Amplification, Library Preparation and Sequencing
2.3. Metagenomic Data Analysis
2.4. Quantitative RT-PCR Assay
2.5. Electron Microscopy
3. Results
3.1. Identification of Known and Novel Viruses by Metagenomic Analysis
3.2. Discovery of Three Distinct Viruses in One Mosquito Pool, 16-0052
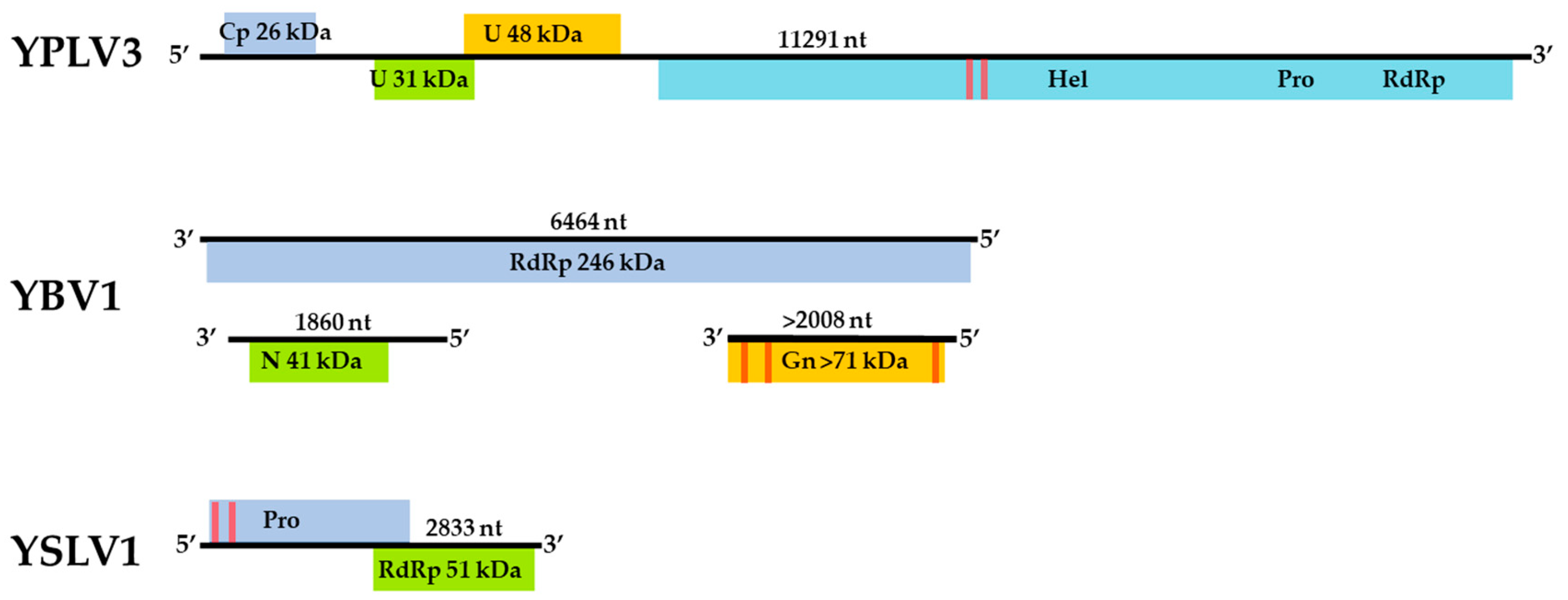
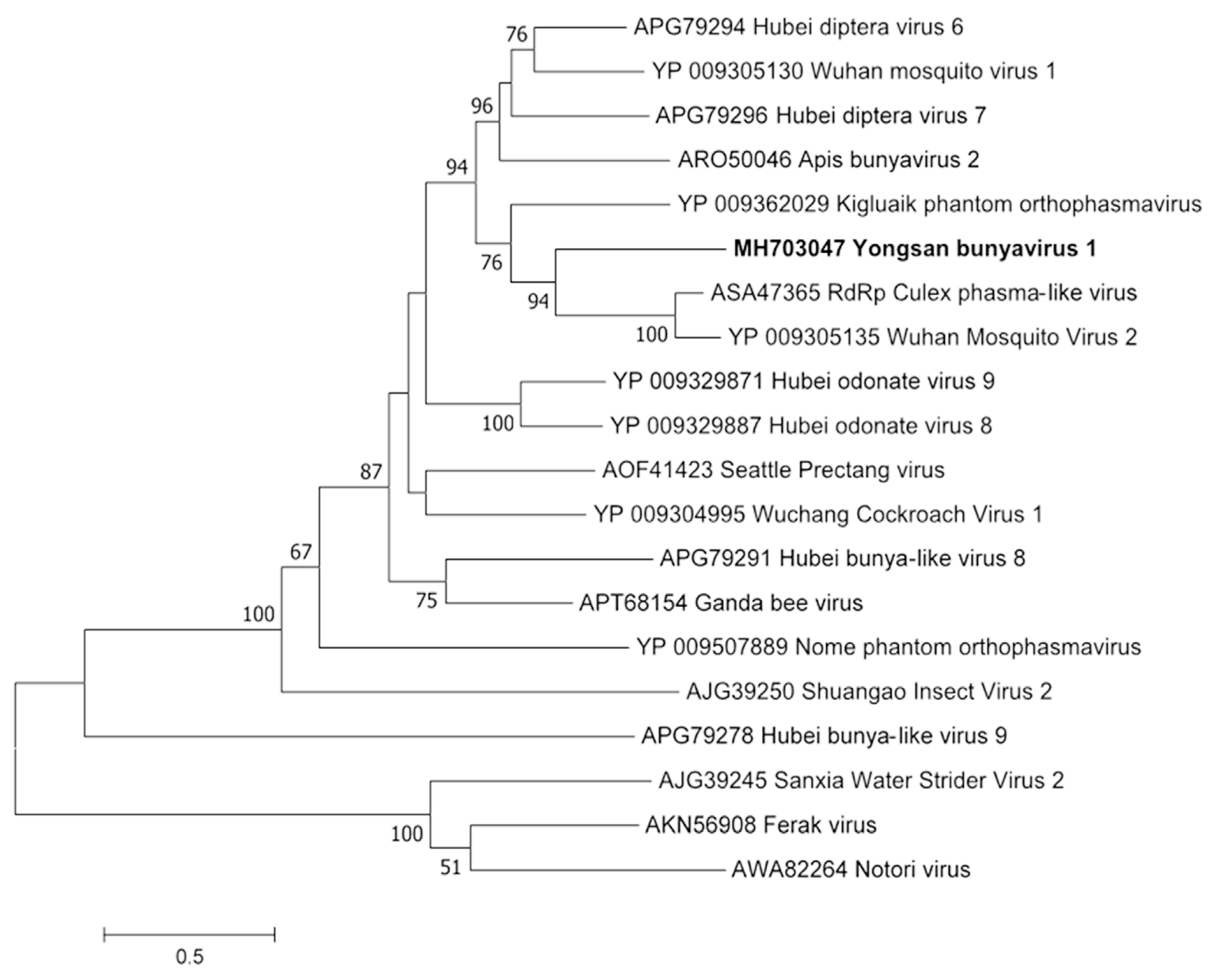
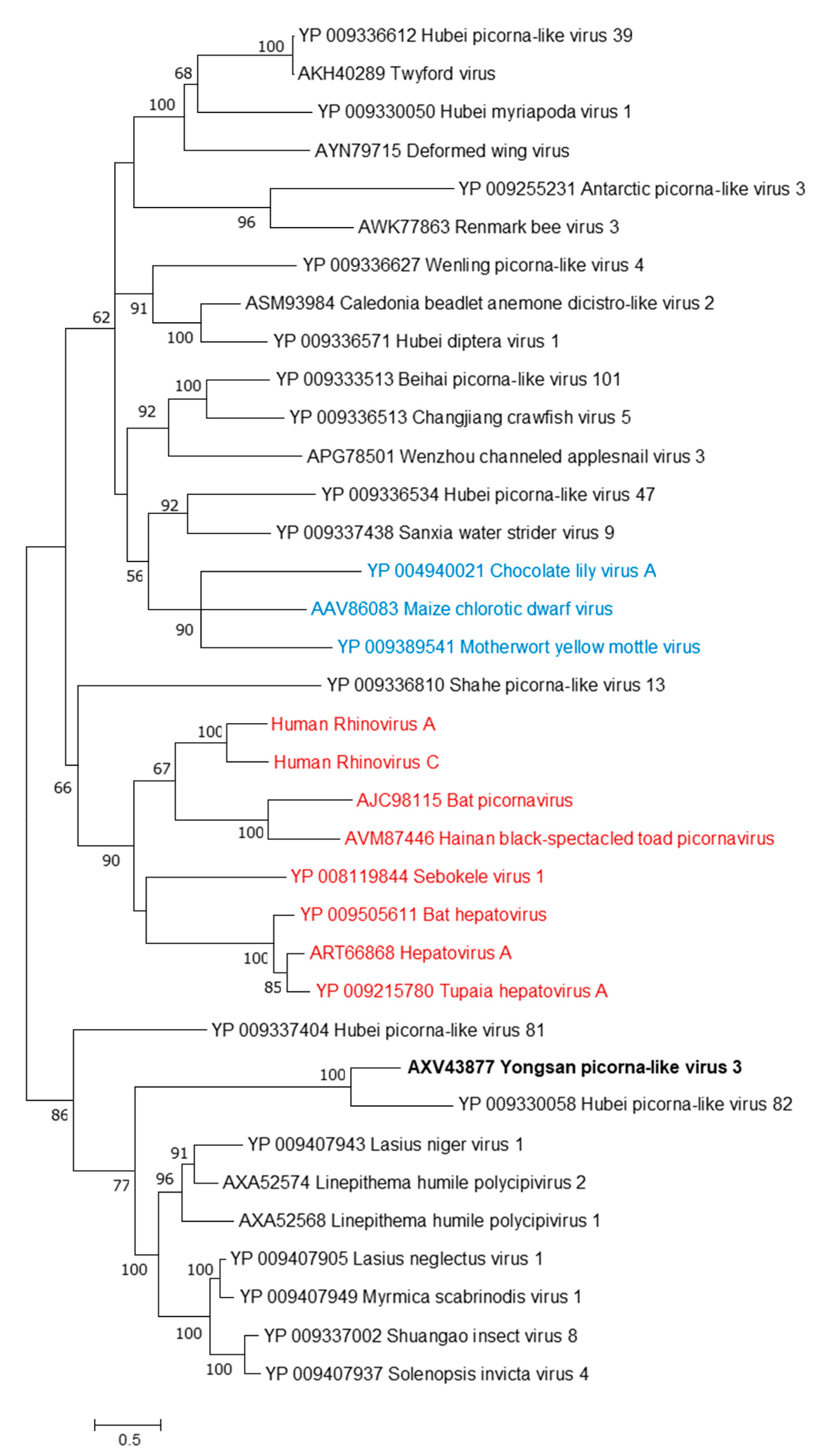
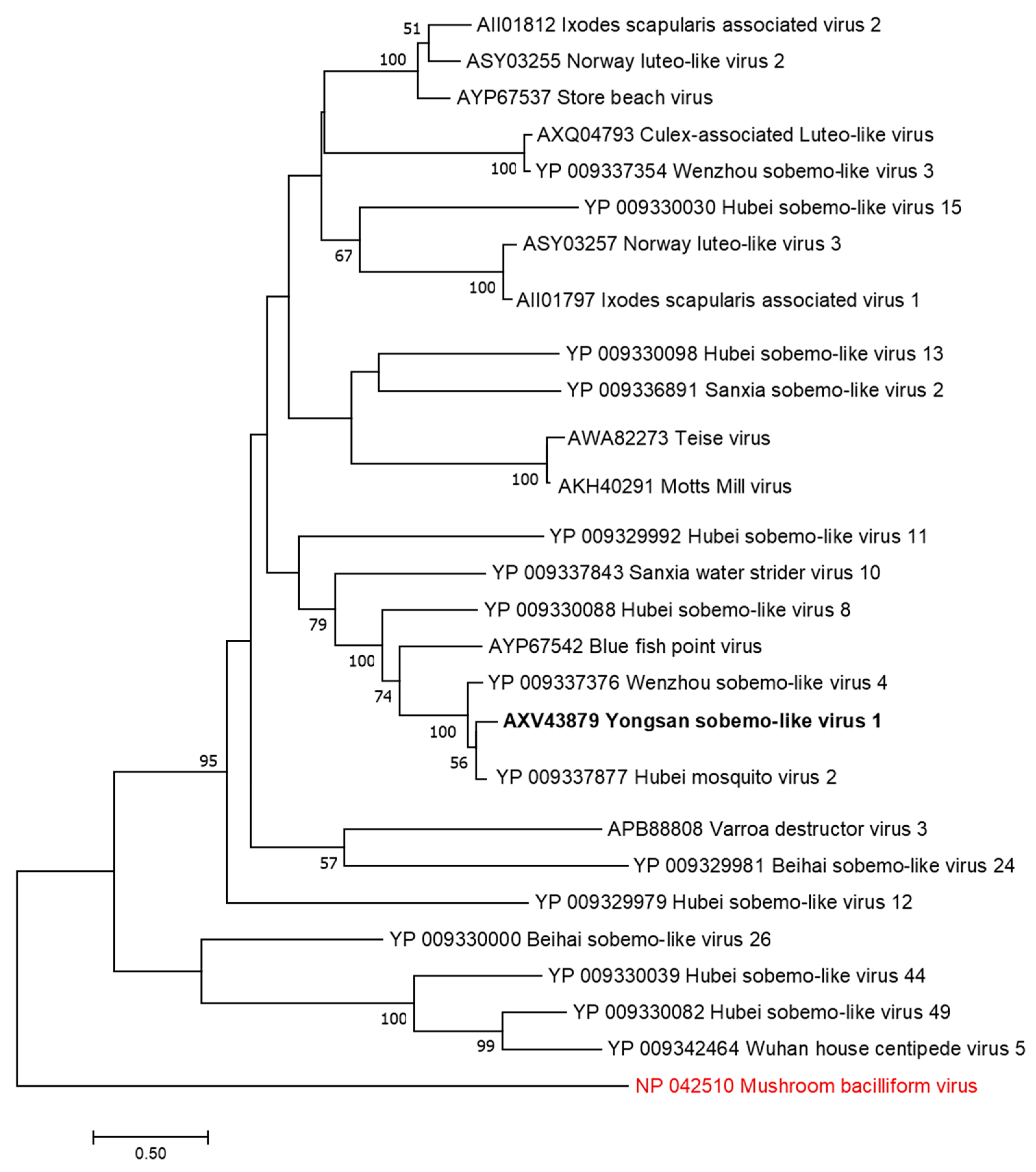
3.3. Genome Sequences and Phylogenetic Analysis of the Viruses
3.4. Wide Distribution of the Viruses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hahn, M.B.; Eisen, R.J.; Eisen, L.; Boegler, K.A.; Moore, C.G.; McAllister, J.; Savage, H.M.; Mutebi, J.P. Reported Distribution of Aedes (Stegomyia) aegypti and Aedes (Stegomyia) albopictus in the United States, 1995–2016 (Diptera: Culicidae). J. Med. Entomol. 2016, 53, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Bolling, B.G.; Weaver, S.C.; Tesh, R.B.; Vasilakis, N. Insect-Specific Virus Discovery: Significance for the Arbovirus Community. Viruses 2015, 7, 4911–4928. [Google Scholar] [CrossRef] [PubMed]
- Napp, S.; Petric, D.; Busquets, N. West Nile virus and other mosquito-borne viruses present in Eastern Europe. Pathog. Glob. Health 2018, 112, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Sukhralia, S.; Verma, M.; Gopirajan, S.; Dhanaraj, P.S.; Lal, R.; Mehla, N.; Kant, C.R. From dengue to Zika: The wide spread of mosquito-borne arboviruses. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Paixao, E.S.; Teixeira, M.G.; Rodrigues, L.C. Zika, chikungunya and dengue: The causes and threats of new and re-emerging arboviral diseases. BMJ Glob. Health 2018, 3, e000530. [Google Scholar] [CrossRef] [PubMed]
- Barzon, L. Ongoing and emerging arbovirus threats in Europe. J. Clin. Virol. 2018, 107, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Papa, A. Emerging arboviral human diseases in Southern Europe. J. Med. Virol. 2017, 89, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, N.M. Challenges and opportunities in controlling mosquito-borne infections. Nature 2018, 559, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.; Pettersson, J.; Higgs, S.; Charrel, R.; de Lamballerie, X. Emerging arboviruses: Why today? One Health 2017, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mayer, S.V.; Tesh, R.B.; Vasilakis, N. The emergence of arthropod-borne viral diseases: A global prospective on dengue, chikungunya and zika fevers. Acta Trop. 2017, 166, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Korkusol, A.; Takhampunya, R.; Hang, J.; Jarman, R.G.; Tippayachai, B.; Kim, H.C.; Chong, S.T.; Davidson, S.A.; Klein, T.A. A novel flavivirus detected in two Aedes spp. collected near the demilitarized zone of the Republic of Korea. J. Gen. Virol. 2017, 98, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- York, A. Viral evolution: Uncovering the secrets of the RNA virosphere. Nat. Rev. Microbiol. 2016, 15, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Maina, A.N.; Luce-Fedrow, A.; Omulo, S.; Hang, J.; Chan, T.C.; Ade, F.; Jima, D.D.; Ogola, E.; Ge, H.; Breiman, R.F.; et al. Isolation and characterization of a novel Rickettsia species (Rickettsia asembonensis sp. nov.) obtained from cat fleas (Ctenocephalides felis). Int. J. Syst. Evol. Microbiol. 2016, 66, 4512–4517. [Google Scholar] [CrossRef] [PubMed]
- Ladner, J.T.; Wiley, M.R.; Beitzel, B.; Auguste, A.J.; Dupuis, A.P.; Lindquist, M.E.; Sibley, S.D.; Kota, K.P.; Fetterer, D.; Eastwood, G.; et al. A multicomponent animal virus isolated from mosquitoes. Cell Host Microbe 2016, 20, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.A.; Thongsripong, P.; Green, A.; Kittayapong, P.; Wilcox, B.A.; Schroth, G.P.; Kapan, D.D.; Bennett, S.N. Metagenomic shotgun sequencing of a Bunyavirus in wild-caught Aedes aegypti from Thailand informs the evolutionary and genomic history of the Phleboviruses. Virology 2014, 464–465, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Nanfack Minkeu, F.; Vernick, K.D. A Systematic review of the natural virome of Anopheles mosquitoes. Viruses 2018, 10, 222. [Google Scholar] [CrossRef] [PubMed]
- Maina, A.N.; Klein, T.A.; Kim, H.C.; Chong, S.T.; Yang, Y.; Mullins, K.; Jiang, J.; St John, H.; Jarman, R.G.; Hang, J.; et al. Molecular characterization of novel mosquito-borne Rickettsia spp. from mosquitoes collected at the Demilitarized Zone of the Republic of Korea. PLoS ONE 2017, 12, e0188327. [Google Scholar] [CrossRef] [PubMed]
- Hang, J.; Klein, T.A.; Kim, H.C.; Yang, Y.; Jima, D.D.; Richardson, J.H.; Jarman, R.G. Genome sequences of five arboviruses in field-captured mosquitoes in a unique rural environment of South Korea. Genome Announc. 2016, 4, e0144-15. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Takhampunya, R.; Tippayachai, B.; Chong, S.T.; Park, J.Y.; Kim, M.S.; Seo, H.J.; Yeh, J.Y.; Lee, W.J.; Lee, D.K.; et al. Japanese encephalitis virus in culicine mosquitoes (Diptera: Culicidae) of the republic of Korea, 2008–2010. Mil. Med. 2015, 180, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Grubaugh, N.D.; Kondig, J.P.; Turell, M.J.; Kim, H.C.; Klein, T.A.; O’Guinn, M.L. Isolation and genomic characterization of Chaoyang virus strain ROK144 from Aedes vexans nipponii from the Republic of Korea. Virology 2013, 435, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Kim, M.S.; Choi, K.S.; Hwang, D.U.; Johnson, J.L.; Klein, T.A. Comparison of adult mosquito black-light and light-emitting diode traps at three cowsheds located in malaria-endemic areas of the Republic of Korea. J. Med. Entomol. 2017, 54, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Hang, J.; Forshey, B.M.; Kochel, T.J.; Li, T.; Solorzano, V.F.; Halsey, E.S.; Kuschner, R.A. Random amplification and pyrosequencing for identification of novel viral genome sequences. J. Biomol. Tech. 2012, 23, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Kilianski, A.; Carcel, P.; Yao, S.; Roth, P.; Schulte, J.; Donarum, G.B.; Fochler, E.T.; Hill, J.M.; Liem, A.T.; Wiley, M.R.; et al. Pathosphere.org: Pathogen detection and characterization through a web-based, open source informatics platform. BMC Bioinform. 2015, 16, 416. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, S.; Raymond, F.; Godzaridis, E.; Laviolette, F.; Corbeil, J. Ray Meta: Scalable de novo metagenome assembly and profiling. Genome Biol. 2012, 13, R122. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2001, 29, 24. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.Y.; Dosztanyi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Biggerstaff, B. PooledInfRate software. Vector Borne Zoonotic Dis. 2005, 5, 420–421. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 4, e05278. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.; Adams, M.J.; Davison, A.J.; Siddell, S.G.; Simmonds, P. (Eds.) Virus Taxonomy: Classification and Nomenclature of Viruses: Online Report of the International Committee on Taxonomy of Viruses. Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/ (accessed on 10 September 2018).
- Olendraite, I.; Lukhovitskaya, N.I.; Porter, S.D.; Valles, S.M.; Firth, A.E. Polycipiviridae: A proposed new family of polycistronic picorna-like RNA viruses. J. Gen. Virol. 2017, 98, 2368–2378. [Google Scholar] [CrossRef] [PubMed]
- Ebi, K.L.; Nealon, J. Dengue in a changing climate. Environ. Res. 2016, 151, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Baylis, M. Potential impact of climate change on emerging vector-borne and other infections in the UK. Environ. Health 2017, 16, 112. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Takhampunya, R.; Kim, H.C.; Tippayachai, B.; Lee, D.K.; Lee, W.J.; Chong, S.T.; Kim, M.S.; Lee, J.S.; Klein, T.A. Distribution and mosquito hosts of Chaoyang virus, a newly reported flavivirus from the Republic of Korea, 2008–2011. J. Med. Entomol. 2014, 51, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Pukhovskaya, N.M.; Morozova, O.V.; Vysochina, N.P.; Belozerova, N.B.; Bakhmetyeva, S.V.; Zdanovskaya, N.I.; Seligman, S.J.; Ivanov, L.I. Tick-borne encephalitis virus in arthropod vectors in the Far East of Russia. Ticks Tick Borne Dis. 2018, 9, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Kim, H.C.; Chong, S.T.; Klein, T.A.; Lee, W.J. Molecular survey of Dirofilaria immitis and Dirofilaria repens by direct PCR for wild caught mosquitoes in the Republic of Korea. Vet. Parasitol. 2007, 148, 149–155. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.; Carstens, E.B.; Lefkowitz, E.J. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier Academic Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Ergonul, O. Crimean–Congo hemorrhagic fever virus: New outbreaks, new discoveries. Curr. Opin. Virol. 2012, 2, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Frey, K.G.; Biser, T.; Hamilton, T.; Santos, C.J.; Pimentel, G.; Mokashi, V.P.; Bishop-Lilly, K.A. Bioinformatic Characterization of Mosquito Viromes within the Eastern United States and Puerto Rico: Discovery of Novel Viruses. Evol. Bioinform. Online 2016, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Junglen, S.; Drosten, C. Virus discovery and recent insights into virus diversity in arthropods. Curr. Opin. Microbiol. 2013, 16, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Junglen, S.; Kurth, A.; Kuehl, H.; Quan, P.-L.; Ellerbrok, H.; Pauli, G.; Nitsche, A.; Nunn, C.; Rich, S.M.; Lipkin, W.I.; et al. Examining Landscape Factors Influencing Relative Distribution of Mosquito Genera and Frequency of Virus Infection. EcoHealth 2009, 6, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Cholleti, H.; Hayer, J.; Fafetine, J.; Berg, M.; Blomstrom, A.L. Genetic characterization of a novel picorna-like virus in Culex spp. mosquitoes from Mozambique. Virol. J. 2018, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Somera, M.; Sarmiento, C.; Truve, E. Overview on Sobemoviruses and a Proposal for the Creation of the Family Sobemoviridae. Viruses 2015, 7, 3076–3115. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.L.; Longdon, B.; Lewis, S.H.; Obbard, D.J. Twenty-Five New Viruses Associated with the Drosophilidae (Diptera). Evol. Bioinform. Online 2016, 12, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Dacheux, L.; Cervantes-Gonzalez, M.; Guigon, G.; Thiberge, J.M.; Vandenbogaert, M.; Maufrais, C.; Caro, V.; Bourhy, H. A preliminary study of viral metagenomics of French bat species in contact with humans: Identification of new mammalian viruses. PLoS ONE 2014, 9, e87194. [Google Scholar] [CrossRef] [PubMed]
- Marklewitz, M.; Zirkel, F.; Kurth, A.; Drosten, C.; Junglen, S. Evolutionary and phenotypic analysis of live virus isolates suggests arthropod origin of a pathogenic RNA virus family. Proc. Natl. Acad. Sci. USA 2015, 112, 7536–7541. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benko, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- DaPalma, T.; Doonan, B.P.; Trager, N.M.; Kasman, L.M. A systematic approach to virus-virus interactions. Virus Res. 2010, 149, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bolling, B.G.; Olea-Popelka, F.J.; Eisen, L.; Moore, C.G.; Blair, C.D. Transmission dynamics of an insect-specific flavivirus in a naturally infected Culex pipiens laboratory colony and effects of co-infection on vector competence for West Nile virus. Virology 2012, 427, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Hobson-Peters, J.; Yam, A.W.; Lu, J.W.; Setoh, Y.X.; May, F.J.; Kurucz, N.; Walsh, S.; Prow, N.A.; Davis, S.S.; Weir, R.; et al. A new insect-specific flavivirus from northern Australia suppresses replication of West Nile virus and Murray Valley encephalitis virus in co-infected mosquito cells. PLoS ONE 2013, 8, e56534. [Google Scholar] [CrossRef] [PubMed]
- Göertz, G.P.; Vogels, C.B.F.; Geertsema, C.; Koenraadt, C.J.M.; Pijlman, G.P. Mosquito co-infection with Zika and chikungunya virus allows simultaneous transmission without affecting vector competence of Aedes aegypti. PLoS Negl. Trop. Dis. 2017, 11, e0005654. [Google Scholar] [CrossRef] [PubMed]
- Newman, C.M.; Cerutti, F.; Anderson, T.K.; Hamer, G.L.; Walker, E.D.; Kitron, U.D.; Ruiz, M.O.; Brawn, J.D.; Goldberg, T.L. Culex flavivirus and West Nile virus mosquito coinfection and positive ecological association in Chicago, United States. Vector Borne Zoonotic Dis. 2011, 11, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.A.; Iturbe-Ormaetxe, I.; Jeffery, J.A.; Lu, G.; Pyke, A.T.; Hedges, L.M.; Rocha, B.C.; Hall-Mendelin, S.; Day, A.; Riegler, M.; et al. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, chikungunya, and Plasmodium. Cell 2009, 139, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.A.; Montgomery, B.L.; Popovici, J.; Iturbe-Ormaetxe, I.; Johnson, P.H.; Muzzi, F.; Greenfield, M.; Durkan, M.; Leong, Y.S.; Dong, Y.; et al. Successful establishment of Wolbachia in Aedes populations to suppress dengue transmission. Nature 2011, 476, 454–457. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Positive Pools | ||||||
|---|---|---|---|---|---|---|
| Species | Number of Mosquitoes in the Pools | Number of Pools Sequenced | YPLV3 | YBV1 | YSLV1 | Total |
| Culex pipiens | 1942 | 120 | 1 | 1 | 2 | 2 |
| Culex tritaeniorhynchus | 1349 | 101 | 1 | 1 | ||
| Aedes vexans nipponii | 1096 | 70 | 2 | 19 | 23 | 32 |
| Aedes albopictus | 553 | 80 | ||||
| Mansonia uniformis | 498 | 76 | ||||
| Coquillettidia ochracea | 228 | 45 | ||||
| Ochlerotatus koreicus | 221 | 67 | 1 | 1 | 2 | |
| Culex inatomii | 123 | 46 | 1 | 1 | ||
| Culex orientalis | 105 | 35 | 1 | 1 | ||
| Culex vagans | 101 | 14 | 1 | 1 | ||
| Culex bitaeniorhynchus | 88 | 57 | ||||
| Culiseta nipponica | 42 | 8 | ||||
| Armigeres subalbatus | 17 | 12 | ||||
| Ochlerotatus dorsalis | 3 | 3 | ||||
| Aedes lineatopennis | 1 | 1 | ||||
| Culex sasai | 1 | 1 | ||||
| Total | 6368 | 736 | 3 | 23 | 28 | 40 |
| Sample Location | No. of Mosquitoes | No. of Pools | YBV1 | YSLV1 | ||
|---|---|---|---|---|---|---|
| Positive Pools | Infection Rate (%) | Positive Pools | Infection Rate (%) | |||
| Daegu | 22 | 14 | 3 | 13.6 | 8 | 40.3 |
| Pyeongtaek | 1740 | 118 | 81 | 8.7 | 72 | 6.9 |
| Gwangju | 332 | 36 | 22 | 10.0 | 16 | 5.9 |
| Seoul | 1399 | 89 | 61 | 8.1 | 63 | 9.6 |
| Total | 3493 | 257 | 167 | 8.8 | 159 | 8.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanborn, M.A.; Klein, T.A.; Kim, H.-C.; Fung, C.K.; Figueroa, K.L.; Yang, Y.; Asafo-adjei, E.A.; Jarman, R.G.; Hang, J. Metagenomic Analysis Reveals Three Novel and Prevalent Mosquito Viruses from a Single Pool of Aedes vexans nipponii Collected in the Republic of Korea. Viruses 2019, 11, 222. https://doi.org/10.3390/v11030222
Sanborn MA, Klein TA, Kim H-C, Fung CK, Figueroa KL, Yang Y, Asafo-adjei EA, Jarman RG, Hang J. Metagenomic Analysis Reveals Three Novel and Prevalent Mosquito Viruses from a Single Pool of Aedes vexans nipponii Collected in the Republic of Korea. Viruses. 2019; 11(3):222. https://doi.org/10.3390/v11030222
Chicago/Turabian StyleSanborn, Mark A., Terry A. Klein, Heung-Chul Kim, Christian K. Fung, Katherine L. Figueroa, Yu Yang, Edward A. Asafo-adjei, Richard G. Jarman, and Jun Hang. 2019. "Metagenomic Analysis Reveals Three Novel and Prevalent Mosquito Viruses from a Single Pool of Aedes vexans nipponii Collected in the Republic of Korea" Viruses 11, no. 3: 222. https://doi.org/10.3390/v11030222
APA StyleSanborn, M. A., Klein, T. A., Kim, H.-C., Fung, C. K., Figueroa, K. L., Yang, Y., Asafo-adjei, E. A., Jarman, R. G., & Hang, J. (2019). Metagenomic Analysis Reveals Three Novel and Prevalent Mosquito Viruses from a Single Pool of Aedes vexans nipponii Collected in the Republic of Korea. Viruses, 11(3), 222. https://doi.org/10.3390/v11030222