A Needle in A Haystack: Tracing Bivalve-Associated Viruses in High-Throughput Transcriptomic Data



Abstract
:1. Introduction
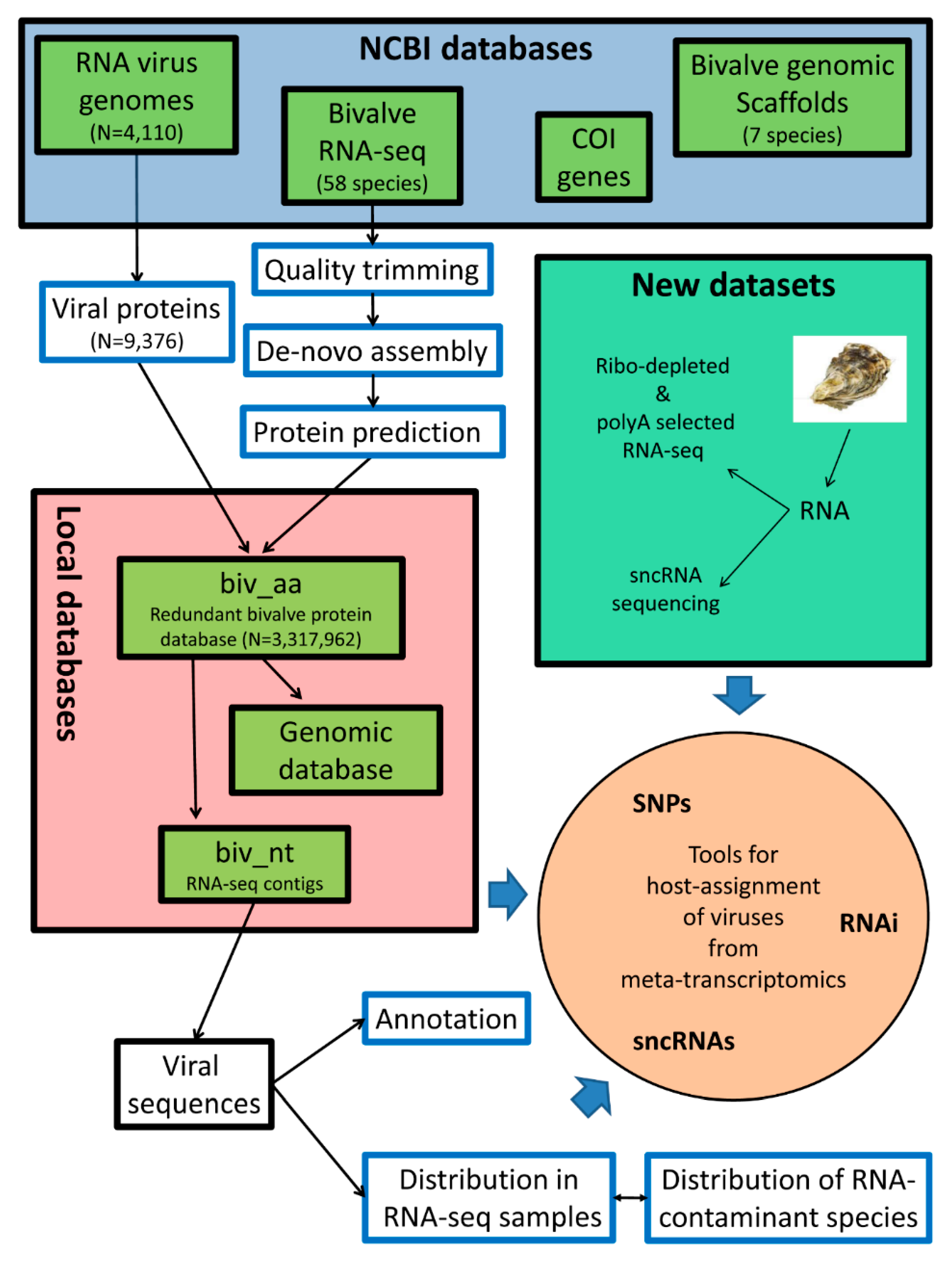
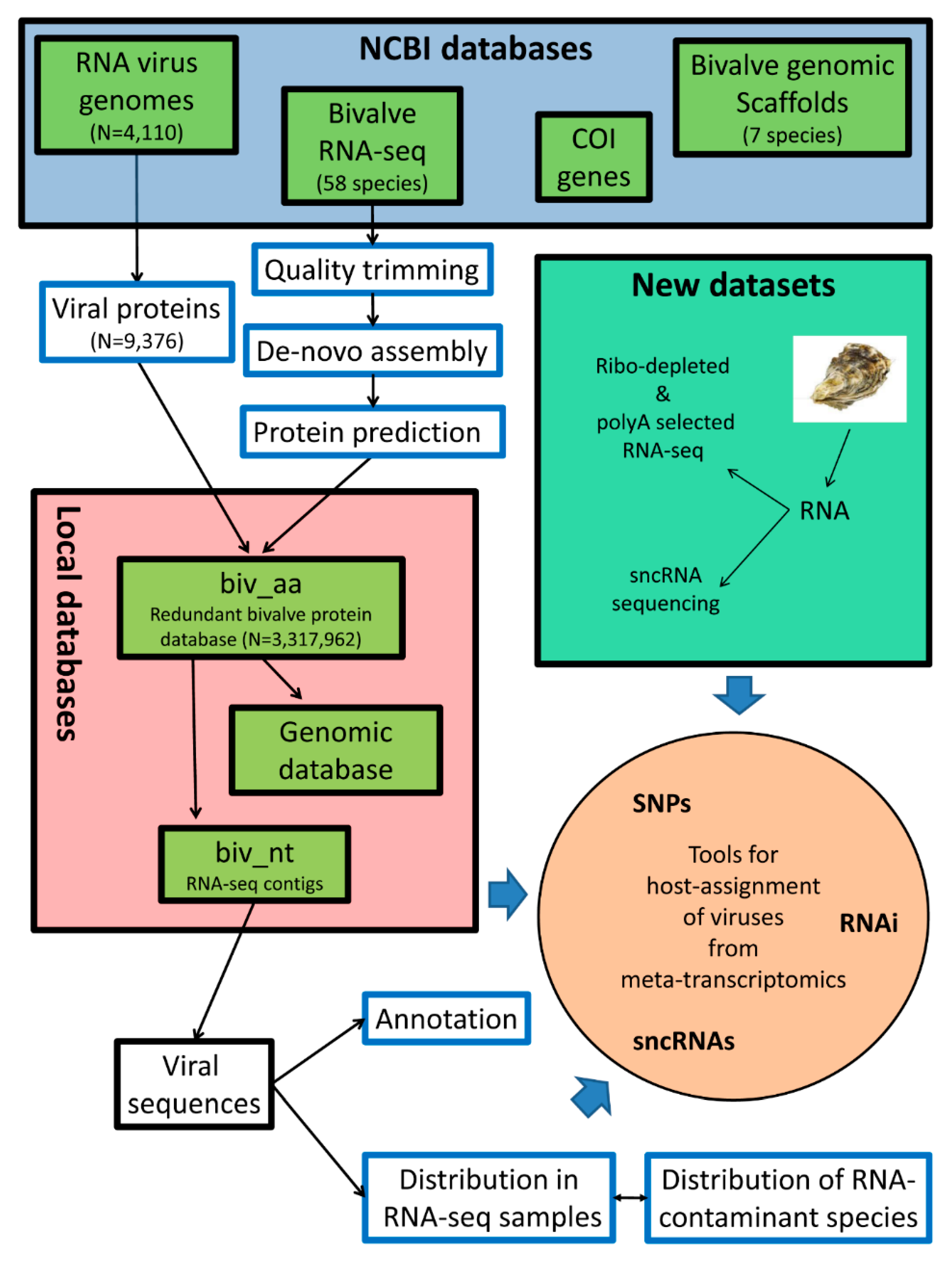
2. Materials and Methods
2.1. Data Retrieval
2.2. Transcriptome de novo Assembly, ORF Prediction, and Protein Domain Mapping
2.3. Identification of Viral Sequences
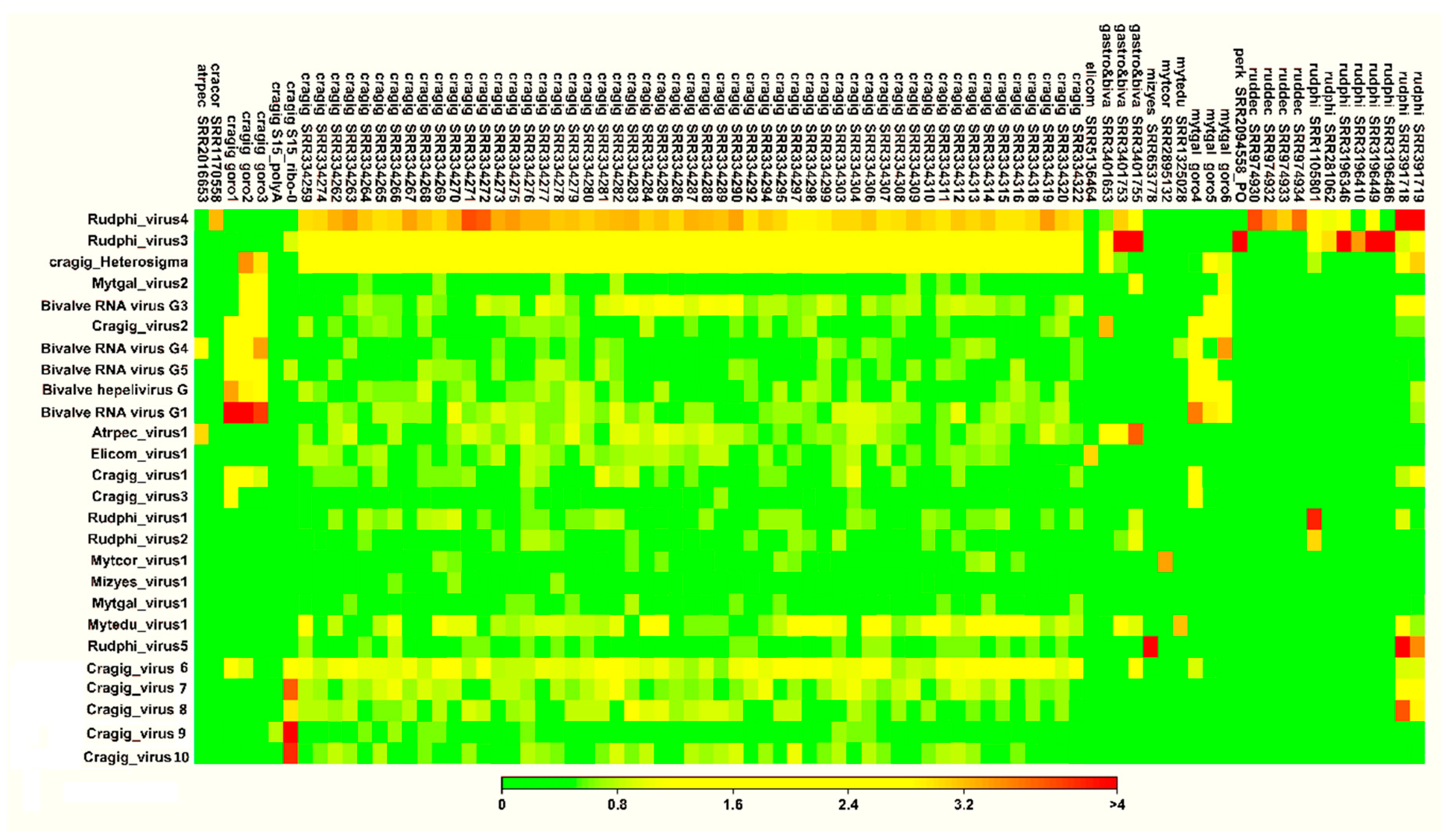
2.4. Distribution of Viruses among RNA-Seq Samples, Expression Analysis, and SNP Calling
2.5. Estimation of the Contamination Levels of RNA-Seq Samples
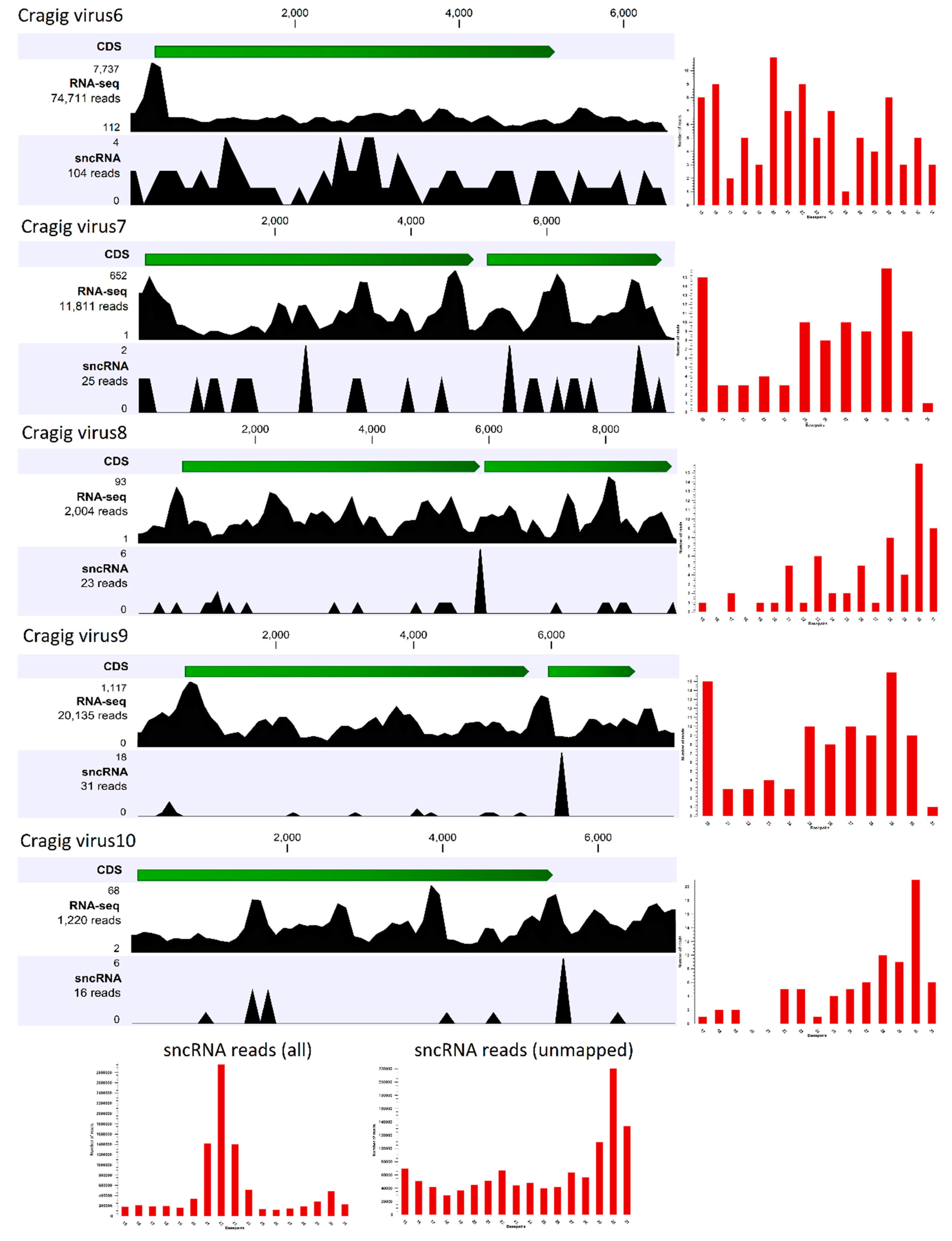
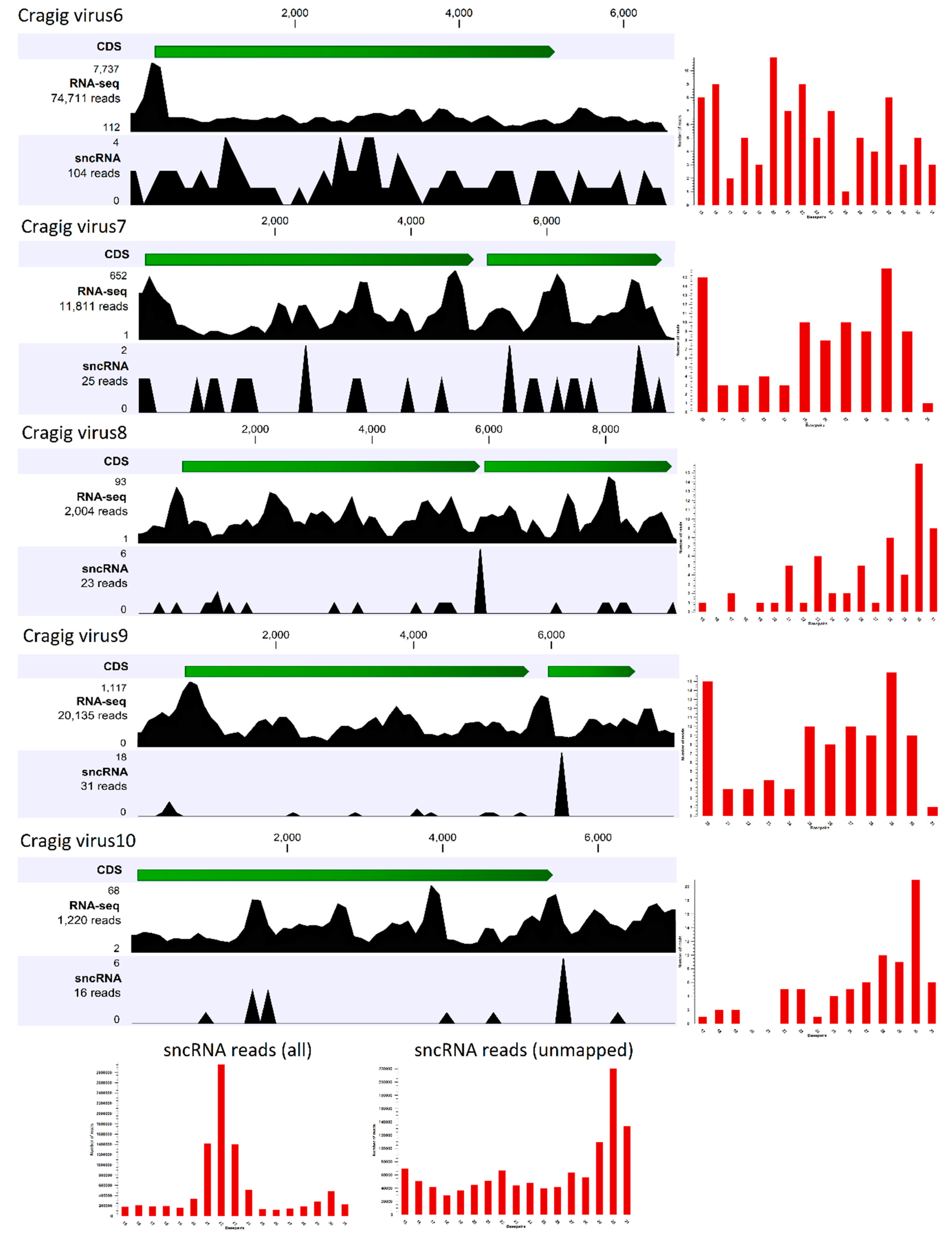
2.6. Small RNA Sequencing and Reads Analysis
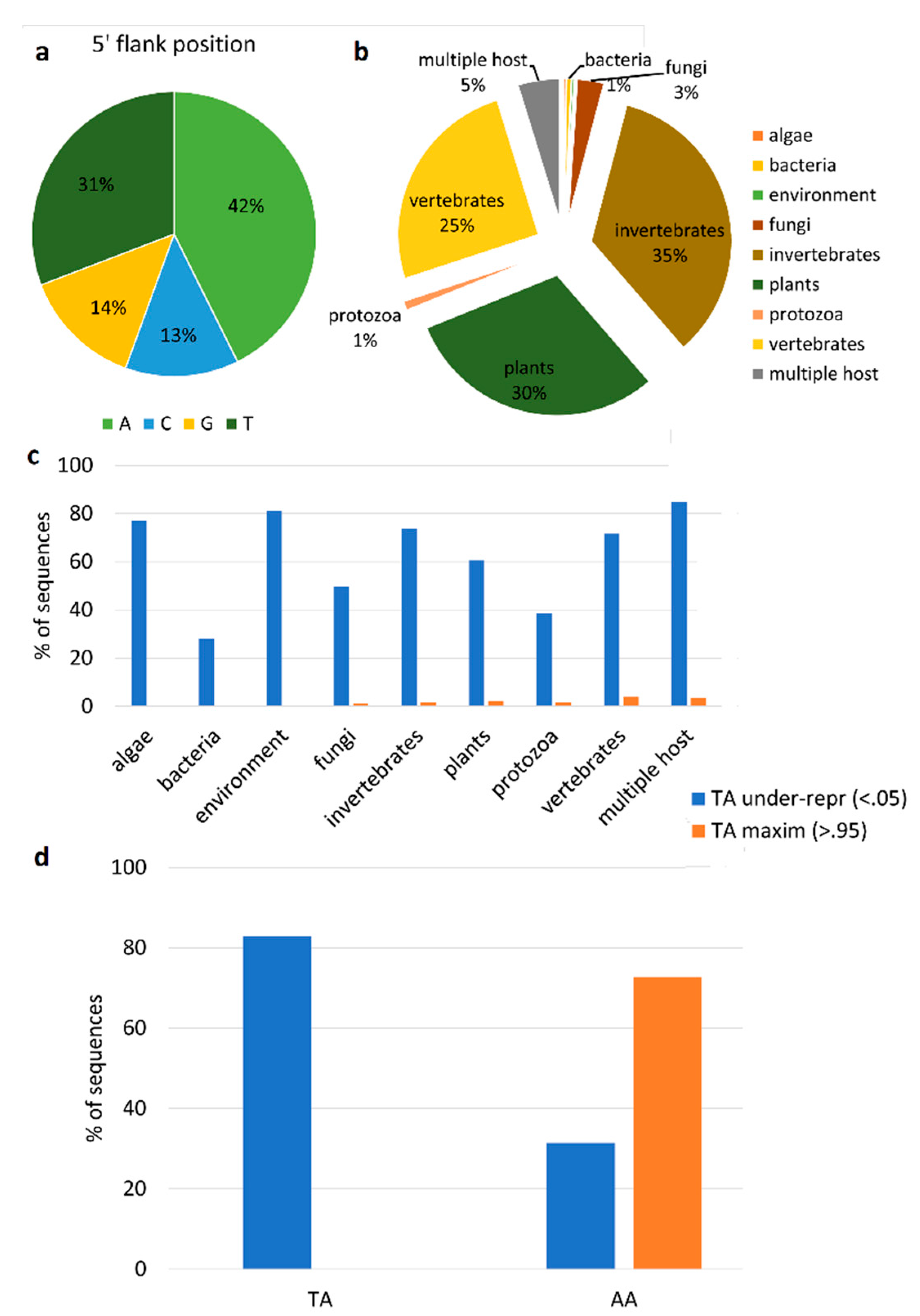
2.7. Analysis of Viral Genome Editing
2.8. Phylogenetic Analysis
3. Results
3.1. Effect of RNA-Seq Library Preparation Protocols on the Detection of Viral Sequences
3.2. Identification of “Nearly Complete” Viral Genomes
3.3. Evaluation of Contaminant RNAs in RNA-Seq Samples
3.4. Tools for the Host-Assignment of Bivalve-Associated Viruses
4. Discussion
Data availability
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses — major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V. Virus world as an evolutionary network of viruses and capsidless selfish elements. Microbiol. Mol. Biol. Rev. MMBR 2014, 78, 278–303. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Yutin, N.; Ivanova, N.N.; Ortega, D.R.; Lee, T.K.; Vierheilig, J.; Daims, H.; Horn, M.; Wagner, M.; Jensen, G.J.; et al. Giant viruses with an expanded complement of translation system components. Science 2017, 356, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Z.; Shi, M.; Holmes, E.C. Using Metagenomics to Characterize an Expanding Virosphere. Cell 2018, 172, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- Mahy, B.W.J. The Evolution and Emergence of RNA Viruses. Emerg. Infect. Dis. 2010, 16, 899. [Google Scholar] [CrossRef]
- Colson, P.; La Scola, B.; Levasseur, A.; Caetano-Anollés, G.; Raoult, D. Mimivirus: Leading the way in the discovery of giant viruses of amoebae. Nat. Rev. Microbiol. 2017, 15, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Duffy, S.; Breitbart, M. A field guide to eukaryotic circular single-stranded DNA viruses: Insights gained from metagenomics. Arch. Virol. 2012, 157, 1851–1871. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, J.; Puigbò, P.; Lobkovsky, A.E.; Wolf, Y.I.; Koonin, E.V. Inevitability of Genetic Parasites. Genome Biol. Evol. 2016, 8, 2856–2869. [Google Scholar] [CrossRef] [PubMed]
- TenOever, B.R. The Evolution of Antiviral Defense Systems. Cell Host Microbe 2016, 19, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. Viruses and mobile elements as drivers of evolutionary transitions. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2016, 371. [Google Scholar] [CrossRef] [PubMed]
- Faillace, C.A.; Lorusso, N.S.; Duffy, S. Overlooking the smallest matter: Viruses impact biological invasions. Ecol. Lett. 2017, 20, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Forterre, P.; Prangishvili, D. The major role of viruses in cellular evolution: Facts and hypotheses. Curr. Opin. Virol. 2013, 3, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; MacCarthy, T. The preferred nucleotide contexts of the AID/APOBEC cytidine deaminases have differential effects when mutating retrotransposon and virus sequences compared to host genes. PLoS Comput. Biol. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M. Virions at the gates: Receptors and the host-virus arms race. PLoS Biol. 2013, 11, e1001574. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Aso, H.; Soper, A.; Yamada, E.; Moriwaki, M.; Juarez-Fernandez, G.; Koyanagi, Y.; Sato, K. A conflict of interest: The evolutionary arms race between mammalian APOBEC3 and lentiviral Vif. Retrovirology 2017, 14, 31. [Google Scholar] [CrossRef] [PubMed]
- Obbard, D.J.; Dudas, G. The genetics of host–virus coevolution in invertebrates. Curr. Opin. Virol. 2014, 8, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Mokili, J.L.; Rohwer, F.; Dutilh, B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2012, 2, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Van Aerle, R.; Santos, E.M. Advances in the application of high-throughput sequencing in invertebrate virology. J. Invertebr. Pathol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Penaud-Budloo, M.; Lecomte, E.; Guy-Duché, A.; Saleun, S.; Roulet, A.; Lopez-Roques, C.; Tournaire, B.; Cogné, B.; Léger, A.; Blouin, V.; et al. Accurate identification and quantification of DNA species by next-generation sequencing in adeno-associated viral vectors produced in insect cells. Hum. Gene Ther. Methods 2017. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L. A decade of RNA virus metagenomics is (not) enough. Virus Res. 2018, 244, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; Wurch, L.L.; Alexander, H.; Dyhrman, S.T.; Gobler, C.J.; Wilhelm, S.W. Virus-host relationships of marine single-celled eukaryotes resolved from metatranscriptomics. Nat. Commun. 2017, 8, 16054. [Google Scholar] [CrossRef] [PubMed]
- Obbard, D.J. Expansion of the metazoan virosphere: Progress, pitfalls, and prospects. Curr. Opin. Virol. 2018, 31, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Bielen, A.; Bošnjak, I.; Sepčić, K.; Jaklič, M.; Cvitanić, M.; Lušić, J.; Lajtner, J.; Simčič, T.; Hudina, S. Differences in tolerance to anthropogenic stress between invasive and native bivalves. Sci. Total Environ. 2016, 543, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Darrigran, G.; Damborenea, C. Ecosystem engineering impact of Limnoperna fortunei in South America. Zoolog. Sci. 2011, 28, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Karatayev, A.Y.; Burlakova, L.E.; Mastitsky, S.E.; Padilla, D.K. Predicting the spread of aquatic invaders: Insight from 200 years of invasion by zebra mussels. Ecol. Appl. Publ. Ecol. Soc. Am. 2015, 25, 430–440. [Google Scholar] [CrossRef]
- Plazzi, F.; Ceregato, A.; Taviani, M.; Passamonti, M. A molecular phylogeny of bivalve mollusks: Ancient radiations and divergences as revealed by mitochondrial genes. PLoS ONE 2011, 6, e27147. [Google Scholar] [CrossRef] [PubMed]
- Faure, B.; Schaeffer, S.W.; Fisher, C.R. Species distribution and population connectivity of deep-sea mussels at hydrocarbon seeps in the Gulf of Mexico. PloS ONE 2015, 10, e0118460. [Google Scholar] [CrossRef] [PubMed]
- Buchmann, K. Evolution of Innate Immunity: Clues from Invertebrates via Fish to Mammals. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Petrone, L.; Kumar, A.; Sutanto, C.N.; Patil, N.J.; Kannan, S.; Palaniappan, A.; Amini, S.; Zappone, B.; Verma, C.; Miserez, A. Mussel adhesion is dictated by time-regulated secretion and molecular conformation of mussel adhesive proteins. Nat. Commun. 2015, 6, 8737. [Google Scholar] [CrossRef] [PubMed]
- Fernández Robledo, J.A.; Yadavalli, R.; Allam, B.; Pales Espinosa, E.; Gerdol, M.; Greco, S.; Stevick, R.J.; Gómez-Chiarri, M.; Zhang, Y.; Heil, C.A.; et al. From the raw bar to the bench: Bivalves as models for human health. Dev. Comp. Immunol. 2019, 92, 260–282. [Google Scholar] [CrossRef] [PubMed]
- Farley, C.A.; Banfield, W.G.; Kasnic, G.; Foster, W.S. Oyster herpes-type virus. Science 1972, 178, 759–760. [Google Scholar] [CrossRef] [PubMed]
- Abbadi, M.; Zamperin, G.; Gastaldelli, M.; Pascoli, F.; Rosani, U.; Milani, A.; Schivo, A.; Rossetti, E.; Turolla, E.; Gennari, L.; et al. Identification of a newly described OsHV-1 µvar from the North Adriatic Sea (Italy). J. Gen. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Wang, C.; Xia, J.; Sun, H.; Zhang, S.; Huang, J. Emerging and endemic types of Ostreid herpesvirus 1 were detected in bivalves in China. J. Invertebr. Pathol. 2015, 124, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Burioli, E. a. V.; Prearo, M.; Riina, M.V.; Bona, M.C.; Fioravanti, M.L.; Arcangeli, G.; Houssin, M. Ostreid herpesvirus type 1 genomic diversity in wild populations of Pacific oyster Crassostrea gigas from Italian coasts. J. Invertebr. Pathol. 2016, 137, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.H.; Kuo, S.T.; Lai, S.H.; Yang, H.S.; Ting, Y.Y.; Hsu, C.L.; Chen, H.C. Herpes-like virus infection causing mortality of cultured abalone Haliotis diversicolor supertexta in Taiwan. Dis. Aquat. Organ. 2005, 65, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Trus, B.L.; Cheng, N.; Steven, A.C.; Watson, M.S.; Cunningham, C.; Le Deuff, R.-M.; Renault, T. A novel class of herpesvirus with bivalve hosts. J. Gen. Virol. 2005, 86, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Martenot, C.; Lethuillier, O.; Fourour, S.; Oden, E.; Trancart, S.; Travaillé, E.; Houssin, M. Detection of undescribed ostreid herpesvirus 1 (OsHV-1) specimens from Pacific oyster, Crassostrea gigas. J. Invertebr. Pathol. 2015, 132, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Arzul, I.; Corbeil, S.; Morga, B.; Renault, T. Viruses infecting marine molluscs. J. Invertebr. Pathol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zannella, C.; Mosca, F.; Mariani, F.; Franci, G.; Folliero, V.; Galdiero, M.; Tiscar, P.G.; Galdiero, M. Microbial Diseases of Bivalve Mollusks: Infections, Immunology and Antimicrobial Defense. Mar. Drugs 2017, 15, 182. [Google Scholar] [CrossRef] [PubMed]
- Renault, T.; Novoa, B. Viruses infecting bivalve molluscs. Aquat. Living Resour. 2004, 17, 397–409. [Google Scholar] [CrossRef]
- Brum, J.R.; Sullivan, M.B. Rising to the challenge: Accelerated pace of discovery transforms marine virology. Nat. Rev. Microbiol. 2015, 13, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Andrade, K.R.; Boratto, P.P.V.M.; Rodrigues, F.P.; Silva, L.C.F.; Dornas, F.P.; Pilotto, M.R.; La Scola, B.; Almeida, G.M.F.; Kroon, E.G.; Abrahão, J.S. Oysters as hot spots for mimivirus isolation. Arch. Virol. 2015, 160, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Iaconelli, M.; Purpari, G.; Della Libera, S.; Petricca, S.; Guercio, A.; Ciccaglione, A.R.; Bruni, R.; Taffon, S.; Equestre, M.; Fratini, M.; et al. Hepatitis A and E Viruses in Wastewaters, in River Waters, and in Bivalve Molluscs in Italy. Food Environ. Virol. 2015, 7, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Suffredini, E.; Proroga, Y.T.R.; Di Pasquale, S.; di Maro, O.; Losardo, M.; Cozzi, L.; Capuano, F.; de Medici, D. Occurrence and Trend of Hepatitis A Virus in Bivalve Molluscs Production Areas Following a Contamination Event. Food Environ. Virol. 2017, 9, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Rosani, U.; Gerdol, M. A bioinformatics approach reveals seven nearly-complete RNA-virus genomes in bivalve RNA-seq data. Virus Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Okinawa Institute of Science and Technology (OIST) Marine Genomics Unit Genome Projects. Available online: http://marinegenomics.oist.jp/ (accessed on 1 January 2019).
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinforma. Oxf. Engl. 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- TrimGalore! Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 1 January 2019).
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinf. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Fang, X.; Guo, X.; Li, L.; Luo, R.; Xu, F.; Yang, P.; Zhang, L.; Wang, X.; Qi, H.; et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 2012, 490, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P.; Kin, K.; Lynch, V.J. A model based criterion for gene expression calls using RNA-seq data. Theory Biosci. Theor. Den Biowiss. 2013, 132, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.-S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.; Meier, S.; MacCarthy, T. The cytidine deaminase under-representation reporter (CDUR) as a tool to study evolution of sequences under deaminase mutational pressure. BMC Bioinf. 2018, 19, 163. [Google Scholar]
- Martinez, T.; Shapiro, M.; Bhaduri-McIntosh, S.; MacCarthy, T. Evolutionary effects of the AID/APOBEC family of mutagenic enzymes on human gamma-herpesviruses. Virus Evol. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Interactive Tree Of Life (iTOL). Available online: https://itol.embl.de/ (accessed on 28 February 2019).
- Venkataraman, S.; Prasad, B.V.L.S.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Dolan, P.T.; Whitfield, Z.J.; Andino, R. Mechanisms and Concepts in RNA Virus Population Dynamics and Evolution. Annu. Rev. Virol. 2018, 5, 69–92. [Google Scholar] [CrossRef] [PubMed]
- Miesen, P.; Joosten, J.; van Rij, R.P. PIWIs Go Viral: Arbovirus-Derived piRNAs in Vector Mosquitoes. PLoS Pathog. 2016, 12, e1006017. [Google Scholar] [CrossRef] [PubMed]
- Vijayendran, D.; Airs, P.M.; Dolezal, K.; Bonning, B.C. Arthropod viruses and small RNAs. J. Invertebr. Pathol. 2013, 114, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Vodovar, N.; Goic, B.; Blanc, H.; Saleh, M.-C. In silico reconstruction of viral genomes from small RNAs improves virus-derived small interfering RNA profiling. J. Virol. 2011, 85, 11016–11021. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 2011, 411, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Interactive Tree Of Life (iTOL). Available online: https://itol.embl.de/shared/umbertorosani (accessed on 28 February 2019).
- Shi, M.; Zhang, Y.-Z.; Holmes, E.C. Meta-transcriptomics and the evolutionary biology of RNA viruses. Virus Res. 2018, 243, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Waldron, F.M.; Stone, G.N.; Obbard, D.J. Metagenomic sequencing suggests a diversity of RNA interference-like responses to viruses across multicellular eukaryotes. PLoS Genet. 2018, 14, e1007533. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-X.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Kang, Y.-J.; Chen, L.-J.; Qin, X.-C.; Xu, J.; Holmes, E.C.; Zhang, Y.-Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, 5378. [Google Scholar] [CrossRef] [PubMed]
- Dolja, V.V.; Koonin, E.V. Metagenomics reshapes the concepts of RNA virus evolution by revealing extensive horizontal virus transfer. Virus Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chin, W.-X.; Ang, S.K.; Chu, J.J.H. Recent advances in therapeutic recruitment of mammalian RNAi and bacterial CRISPR-Cas DNA interference pathways as emerging antiviral strategies. Drug Discov. Today 2017, 22, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-Y.; Chen, C.-C.G.; Conte, D.; Moresco, J.J.; Chaves, D.A.; Mitani, S.; Yates, J.R.; Tsai, M.-D.; Mello, C.C. A ribonuclease coordinates siRNA amplification and mRNA cleavage during RNAi. Cell 2015, 160, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Petit, M.; Mongelli, V.; Frangeul, L.; Blanc, H.; Jiggins, F.; Saleh, M.-C. piRNA pathway is not required for antiviral defense in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2016, 113, E4218–E4227. [Google Scholar] [CrossRef] [PubMed]
- Gammon, D.B.; Ishidate, T.; Li, L.; Gu, W.; Silverman, N.; Mello, C.C. The Antiviral RNA Interference Response Provides Resistance to Lethal Arbovirus Infection and Vertical Transmission in Caenorhabditis elegans. Curr. Biol. CB 2017, 27, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. Evolution of RNA- and DNA-guided antivirus defense systems in prokaryotes and eukaryotes: Common ancestry vs convergence. Biol. Direct 2017, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Green, T.J.; Rolland, J.-L.; Vergnes, A.; Raftos, D.; Montagnani, C. OsHV-1 countermeasures to the Pacific oyster’s anti-viral response. Fish Shellfish Immunol. 2015, 47, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Green, T.J.; Speck, P. Antiviral Defense and Innate Immune Memory in the Oyster. Viruses 2018, 10, 133. [Google Scholar] [CrossRef] [PubMed]
- Bass, B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002, 71, 817–846. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-C.; Liao, W.-Y.; Buckley, K.M.; Yang, S.Y.; Rast, J.P.; Fugmann, S.D. AID/APOBEC-like cytidine deaminases are ancient innate immune mediators in invertebrates. Nat. Commun. 2018, 9, 1948. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.A.; Keegan, L.P.; Wilfert, L.; O’Connell, M.A.; Jiggins, F.M. Evidence for ADAR-induced hypermutation of the Drosophila sigma virus (Rhabdoviridae). BMC Genet. 2009, 10, 75. [Google Scholar] [CrossRef] [PubMed]
- Porath, H.T.; Carmi, S.; Levanon, E.Y. A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat. Commun. 2014, 5, 4726. [Google Scholar] [CrossRef] [PubMed]
- Porath, H.T.; Schaffer, A.A.; Kaniewska, P.; Alon, S.; Eisenberg, E.; Rosenthal, J.; Levanon, E.Y.; Levy, O. A-to-I RNA Editing in the Earliest-Diverging Eumetazoan Phyla. Mol. Biol. Evol. 2017, 34, 1890–1901. [Google Scholar] [CrossRef] [PubMed]
- Piontkivska, H.; Matos, L.F.; Paul, S.; Scharfenberg, B.; Farmerie, W.G.; Miyamoto, M.M.; Wayne, M.L. Role of Host-Driven Mutagenesis in Determining Genome Evolution of Sigma Virus (DMelSV.; Rhabdoviridae) in Drosophila melanogaster. Genome Biol. Evol. 2016, 8, 2952–2963. [Google Scholar] [CrossRef] [PubMed]
- Piontkivska, H.; Frederick, M.; Miyamoto, M.M.; Wayne, M.L. RNA editing by the host ADAR system affects the molecular evolution of the Zika virus. Ecol. Evol. 2017, 7, 4475–4485. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SRA ID | Sample ID | RNA Selection Method | No. of Reads (M) | No. Of Assembled Contig | No. of Predicted Protein | No. of vRdRp | Ratio of Viral Proteins | No. of Complete Genomes * |
|---|---|---|---|---|---|---|---|---|
| SRR3401648 | SAMN04625952 | Ribo-depletion | 99.7 | 96,102 | 45,343 | 30 | 0.00007 | 2 |
| SRR3401653 | polyA-selection | 58.3 | 120,399 | 38,498 | 129 | 0.00034 | 9 | |
| SRR3401753 | SAMN04625958 | Ribo-depletion | 47.9 | 180,272 | 48,687 | 43 | 0.00009 | 6 |
| SRR3401755 | polyA-selection | 60.3 | 105,611 | 14,661 | 54 | 0.00037 | 4 | |
| SRR7637587 | SAMN09760011 | Ribo-depletion | 54.1 | 156,166 | 41,785 | 46 | 0.00011 | 5 |
| SRR8237210 | polyA-selection | 52.0 | 93,172 | 40,301 | 10 | 0.00002 | 0 |
| Species | Viral Sequences | Tissue | Geographic Origin | Library Type | Virus Name | Virus Distribution among Bivalve RNA-seq | Total Viral Reads | % of Viral Reads * | NCBI ID | Blastp | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | nr | Nearly Complete | E-Value | Description | Identity % | |||||||||
| Atrina pectinata | 17 | 15 | 1 | mixed | China | PA | Atrpec_virus1 | \ | 2104 | 0.00202 | MG210792 | 0 | Wenzhou picorna-like virus 26 | 99.57 |
| Crassostrea gigas | 148 | 109 | 13 | gills | Italy | PA | Cragig_virus3 | mytgal | 898 | 0.00135 | MG210795 | \ | \ | \ |
| PA | Bivalve hepelivirus G | mytgal | 4058 | 0.00611 | KX158876 | 0 | Bivalve hepelivirus G | 99.95 | ||||||
| PA | Bivalve RNA virus G5 | \ | 1382 | 0.00173 | KX158874 | 0 | Bivalve RNA virus G5 | 100 | ||||||
| PA | Bivalve RNA virus G3 | mytgal | 1286 | 0.00161 | KX158873 | 0 | Bivalve RNA virus G3 | 100 | ||||||
| PA | Cragig_virus1 | \ | 890 | 0.00134 | MG210793 | \ | \ | \ | ||||||
| PA | Cragig_virus2 | mytgal | 468 | 0.00058 | MG210794 | 0 | Wenzhou picorna-like virus 24 | 93.6 | ||||||
| PA | Bivalve RNA virus G1 | mytgal | 37902 | 0.04732 | KX158871 | 0 | Bivalve RNA virus G1 | 100 | ||||||
| PA | AY337486 | mytgal | 5156 | 0.00644 | AY337486 | 0 | Heterosigma akashiwo RNA virus | 100 | ||||||
| RD | Cragig_virus6 | \ | 30047 | 0.05554 | MK561968 | 1^-108 | Beihai picorna-like virus 21 | 66 | ||||||
| RD | Cragig_virus7 | \ | 5493 | 0.01015 | MK561969 | 3^-40 | Wenzhou picorna-like virus 41 | 70 | ||||||
| RD | Cragig_virus8 | \ | 955 | 0.00177 | MK561970 | 0 | Rhizosolenia setigera RNA virus | 69 | ||||||
| RD | Cragig_virus9 | \ | 9200 | 0.01701 | MK561971 | \ | \ | \ | ||||||
| RD | Cragig_virus10 | \ | 568 | 0.00105 | MK561972 | \ | \ | \ | ||||||
| Elliptio complanata | 2 | 1 | 1 | mixed | USA | PA | Elicom_virus1 | \ | 2268 | 0.00552 | MG210796 | \ | \ | \ |
| Mizuhopecten yessoensis | 25 | 25 | 1 | mixed | China | PA | Mizyes_virus1 | \ | 1974 | 0.00522 | MG210800 | \ | \ | \ |
| Mytilus coruscus | 1 | mixed | China | PA | Mytcor_virus1 | \ | 4460 | 0.01028 | MG210801 | 0 | Pitaya virus X isolate P37 | 98.24 | ||
| Mytilus edulis | 37 | 33 | 1 | mixed | France | PA | Mytedu_virus1 | \ | 1936 | 0.00694 | MG210802 | 0 | Barns Ness breadcrumb sponge aquatic picorna-like virus 2 | 99 |
| Mytilus galloprovincialis | 115 | 52 | 3 | gills | Italy | PA | Bivalve RNA virus G4 | cragig, atrpec | 4818 | 0.00713 | KX158875 | 0 | Bivalve RNA virus G4 | 99.77 |
| PA | Mytgal_virus1 | cragig | \ | \ | MG210803 | \ | \ | \ | ||||||
| PA | Mytgal_virus2 | cragig | 1432 | 0.00212 | MG210804 | 0 | Wenzhou picorna-like virus 51 | 78.7 | ||||||
| Ruditapes philippinarum | 121 | 49 | 5 | gills | China | PA | Rudphi_virus1 | \ | 9842 | 0.02869 | MG210805 | 0 | Wenzhou picorna-like virus 38 | 72.9 |
| gills | China | PA | Rudphi_virus2 | \ | 1031 | 0.00301 | MG210806 | \ | \ | \ | ||||
| gills | China | PA | Rudphi_virus3 | \ | 181992 | 0.29884 | MG210807 | 0 | Wenzhou gastropodes virus 2 | 97.3 | ||||
| gills | China | PA | Rudphi_virus4 | ruddec, ostste, ostlur, cracor, cragig, mytedu | 5388365 | 3.06157 | MG210808 | 0 | Wenzhou gastropodes virus 1 | 92 | ||||
| larvae | USA | PA | Rudphi_virus5 | \ | 19965 | 0.02936 | MG210809 | 0 | Marine RNA virus BC-4 | 70 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosani, U.; Shapiro, M.; Venier, P.; Allam, B. A Needle in A Haystack: Tracing Bivalve-Associated Viruses in High-Throughput Transcriptomic Data. Viruses 2019, 11, 205. https://doi.org/10.3390/v11030205
Rosani U, Shapiro M, Venier P, Allam B. A Needle in A Haystack: Tracing Bivalve-Associated Viruses in High-Throughput Transcriptomic Data. Viruses. 2019; 11(3):205. https://doi.org/10.3390/v11030205
Chicago/Turabian StyleRosani, Umberto, Maxwell Shapiro, Paola Venier, and Bassem Allam. 2019. "A Needle in A Haystack: Tracing Bivalve-Associated Viruses in High-Throughput Transcriptomic Data" Viruses 11, no. 3: 205. https://doi.org/10.3390/v11030205
APA StyleRosani, U., Shapiro, M., Venier, P., & Allam, B. (2019). A Needle in A Haystack: Tracing Bivalve-Associated Viruses in High-Throughput Transcriptomic Data. Viruses, 11(3), 205. https://doi.org/10.3390/v11030205