Protein Disulfide Isomerase Inhibitor Suppresses Viral Replication and Production during Antibody-Dependent Enhancement of Dengue Virus Infection in Human Monocytic Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dengue Viruses and Antibodies
2.2. Dengue Virus Propagation
2.3. Dengue Virus Titration
2.4. Antibody-Dependent Enhancement Assay
2.5. Detection of Cell Viability
2.6. Phosphoprotein Enrichment
2.7. One-Dimensional Polyacrylamide Gel Electrophoresis (1D PAGE)
2.8. In-Gel Tryptic Digestion of 1D-PAGE with Reduction and Alkylation
2.9. Mass Spectrometric Analysis
2.10. Western Blot Analysis
2.11. Functional Analysis of Protein Disulfide Isomerase
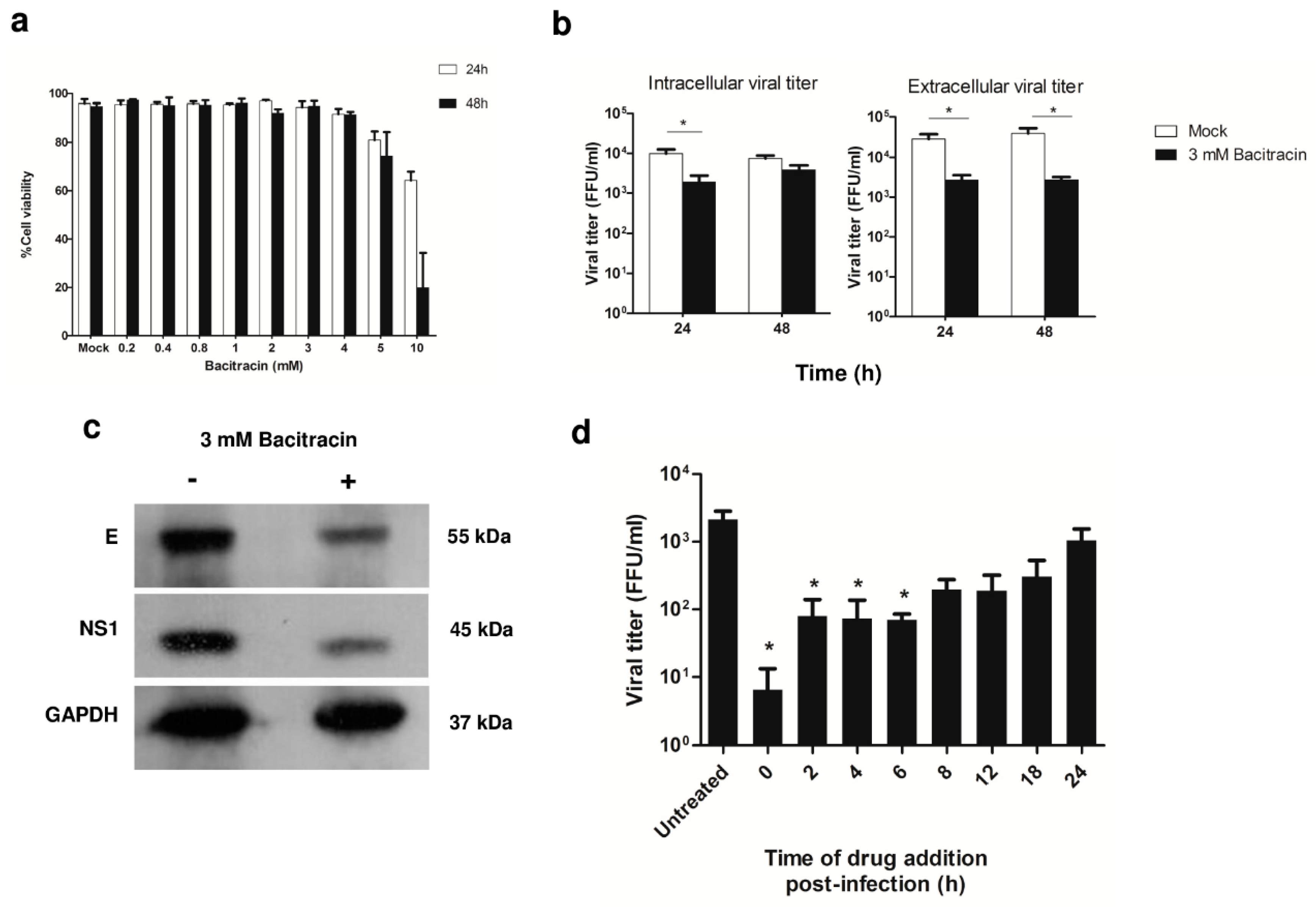
2.12. Time-of-Drug-Addition Assay
2.13. Immunofluorescence Staining
2.14. Data Analysis
3. Results
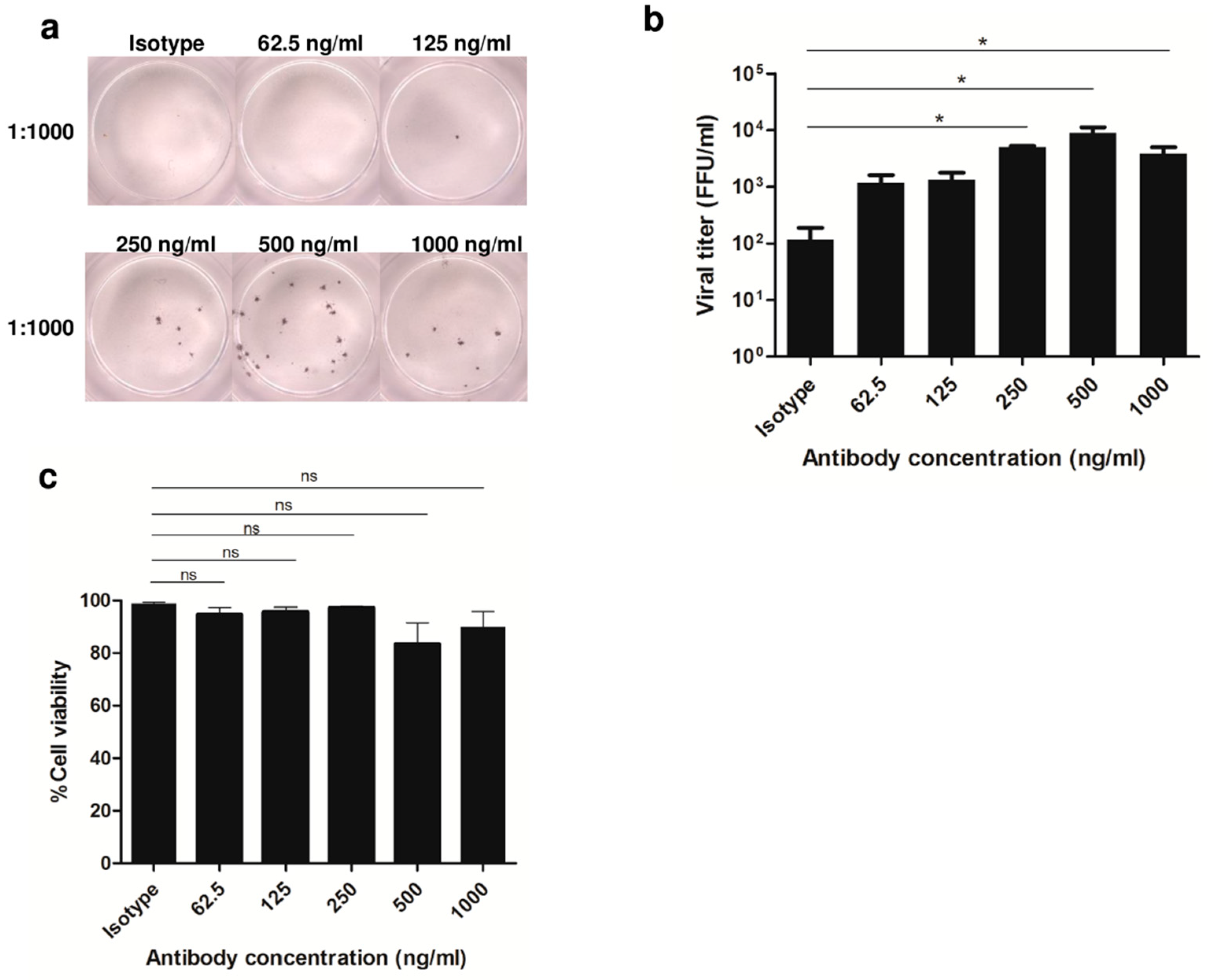
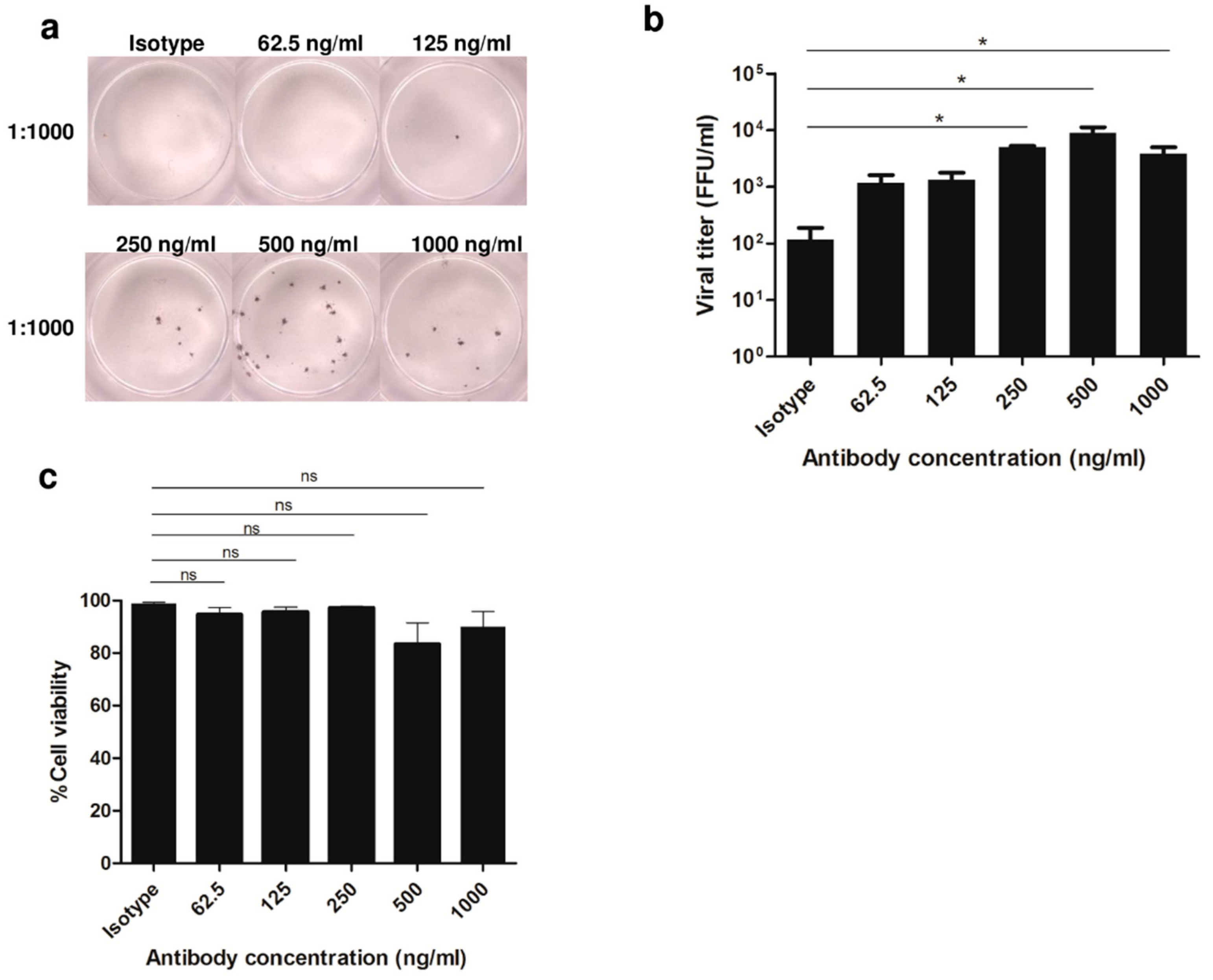
3.1. Enhancement of DENV2 Infection with Cross-Reactive Anti-E Antibody in Human Monocytic Cell Lines
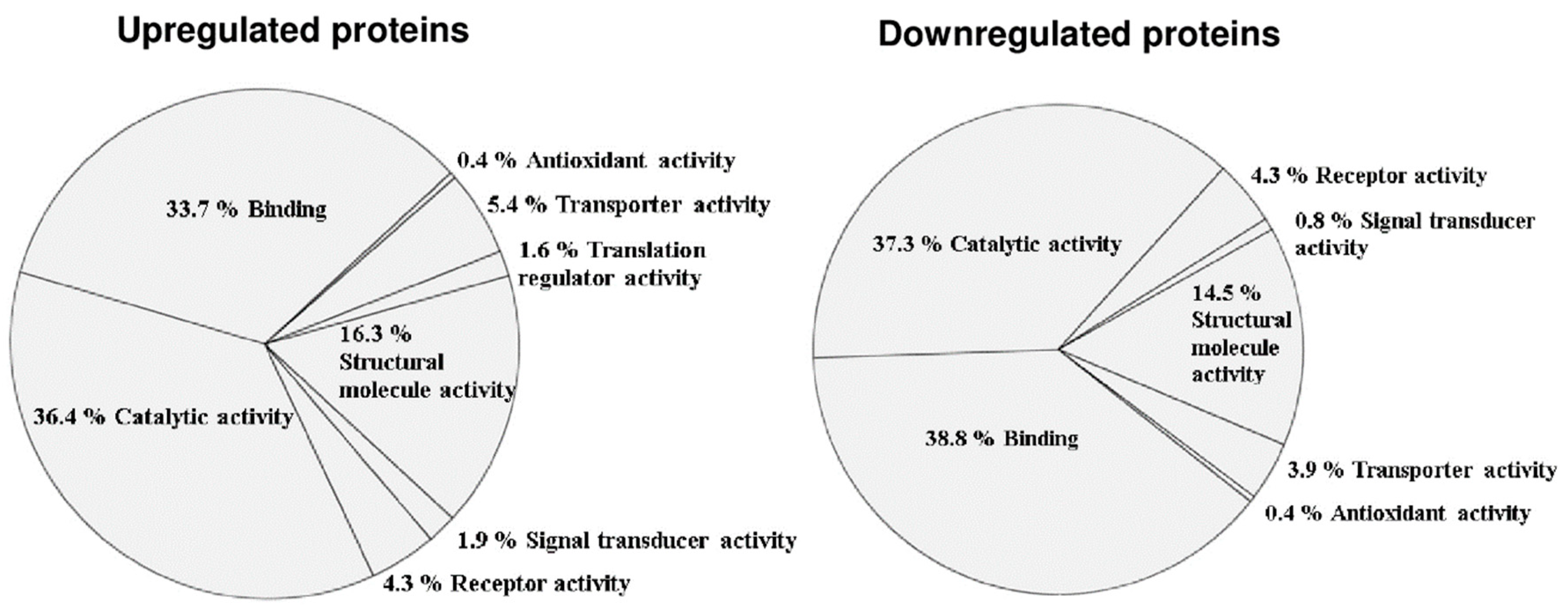
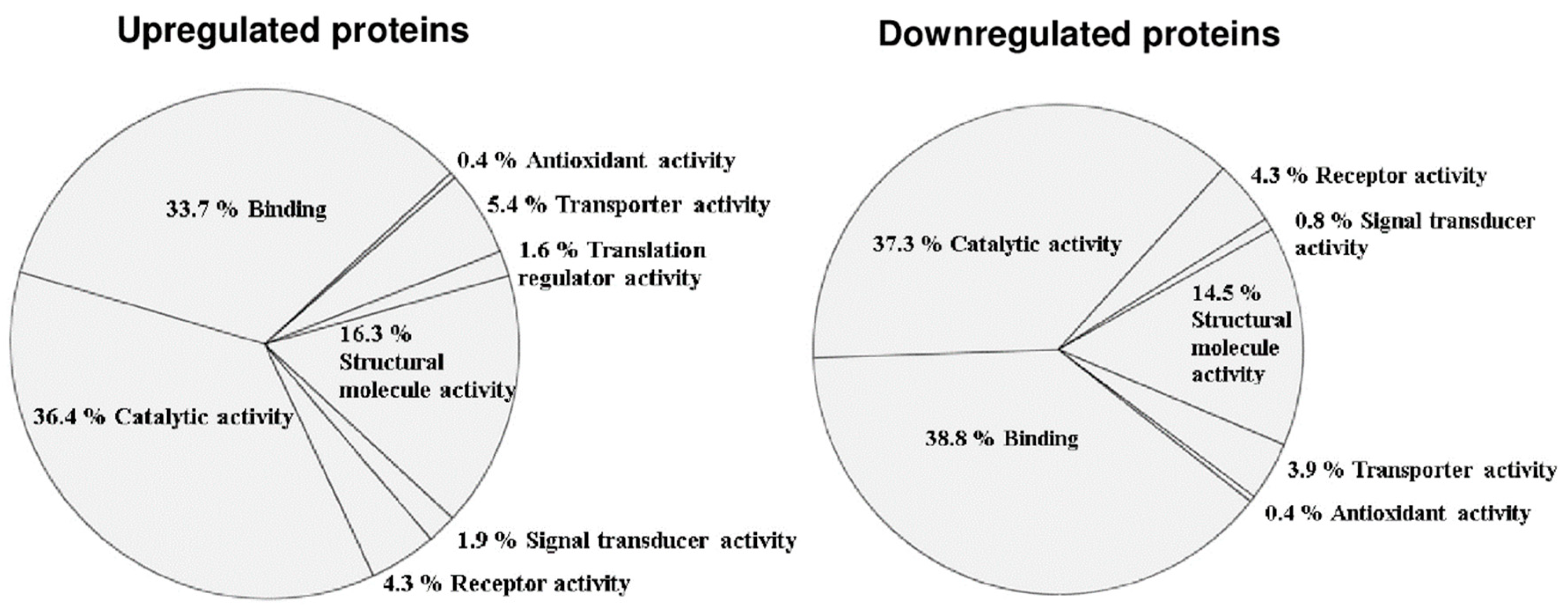
3.2. Altered Phosphoprotein Response to DENV2 Infection in Human Monocytic Cells under ADE Condition
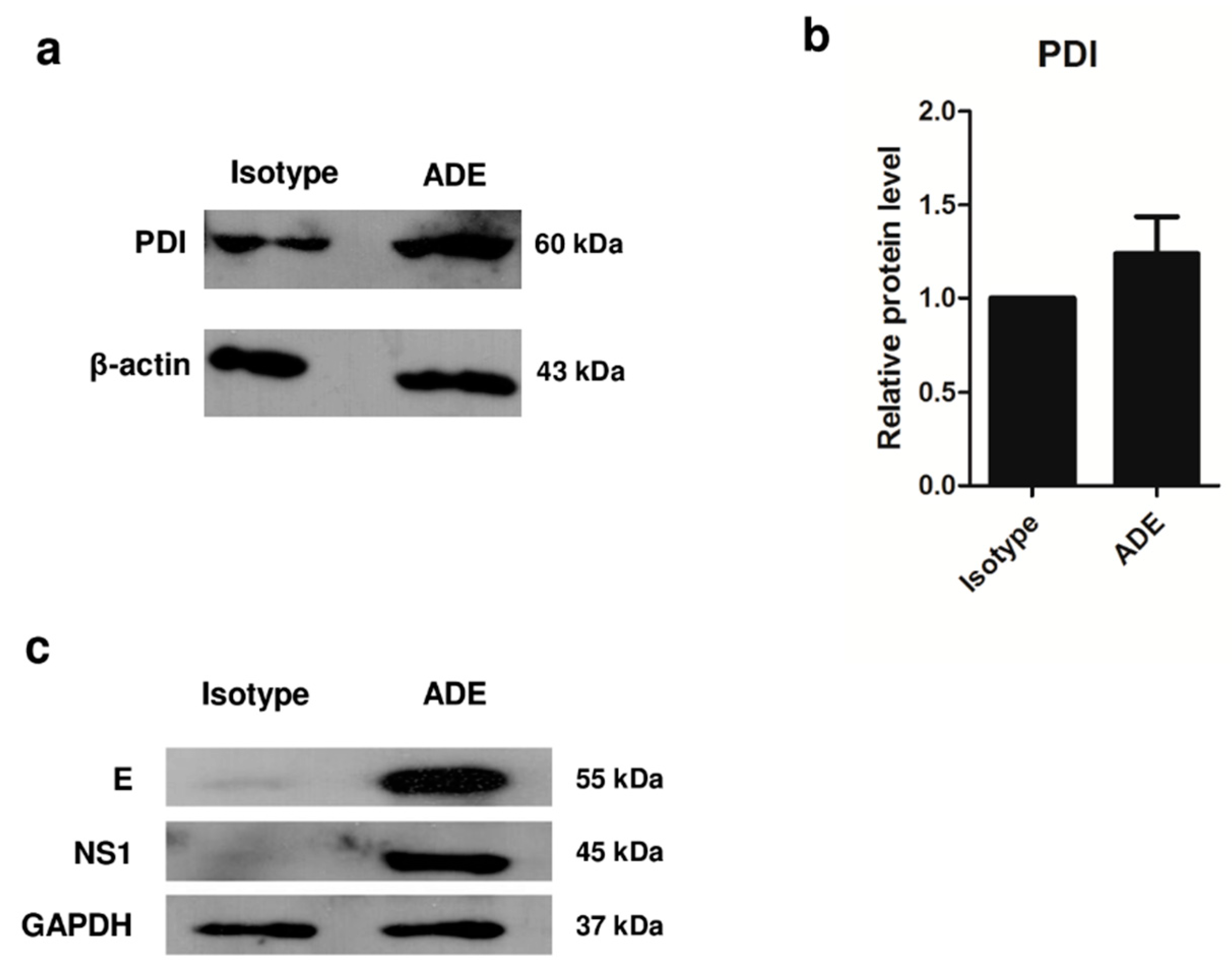
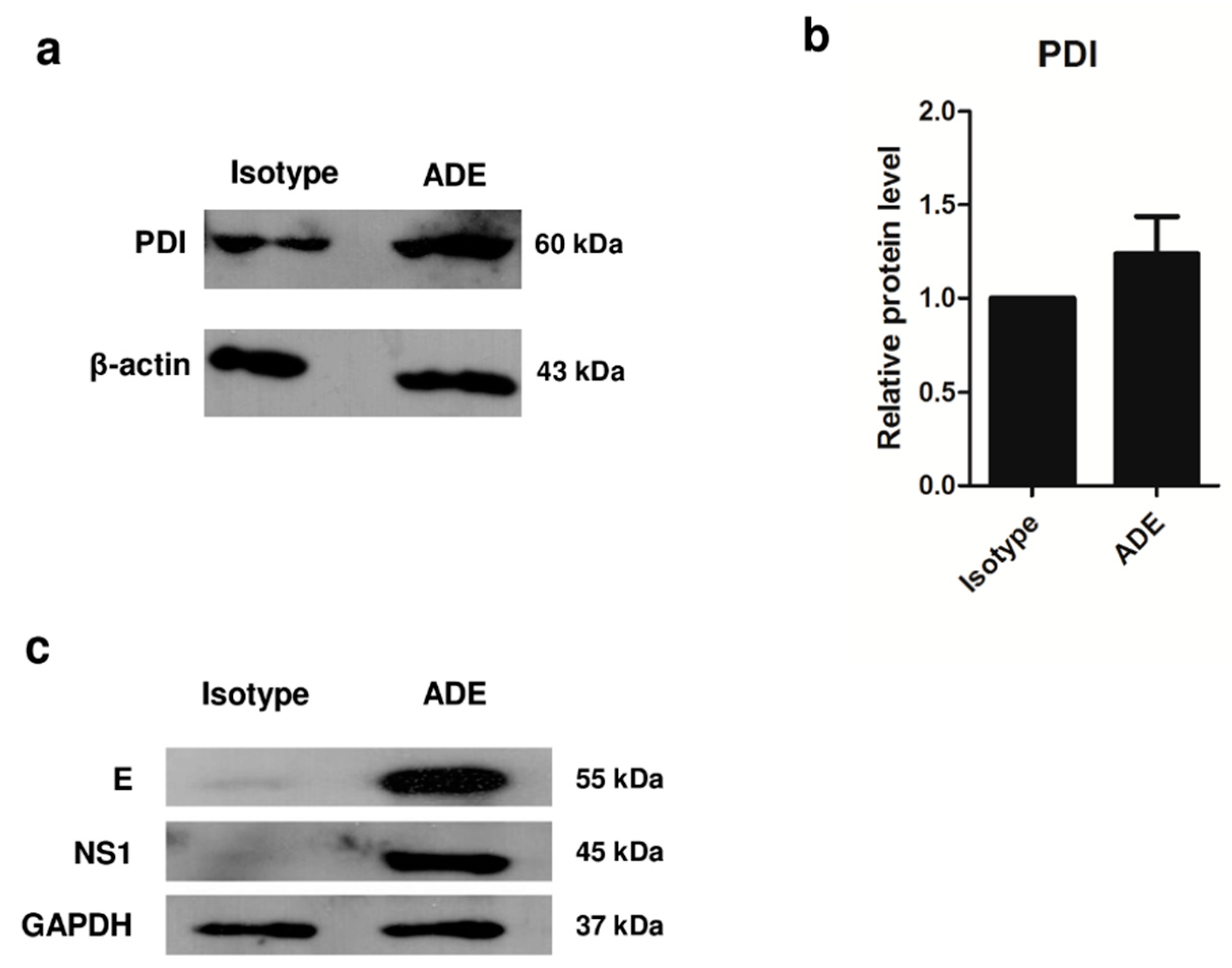
3.3. Validation of Protein Expression by 1D Western Blot Analysis
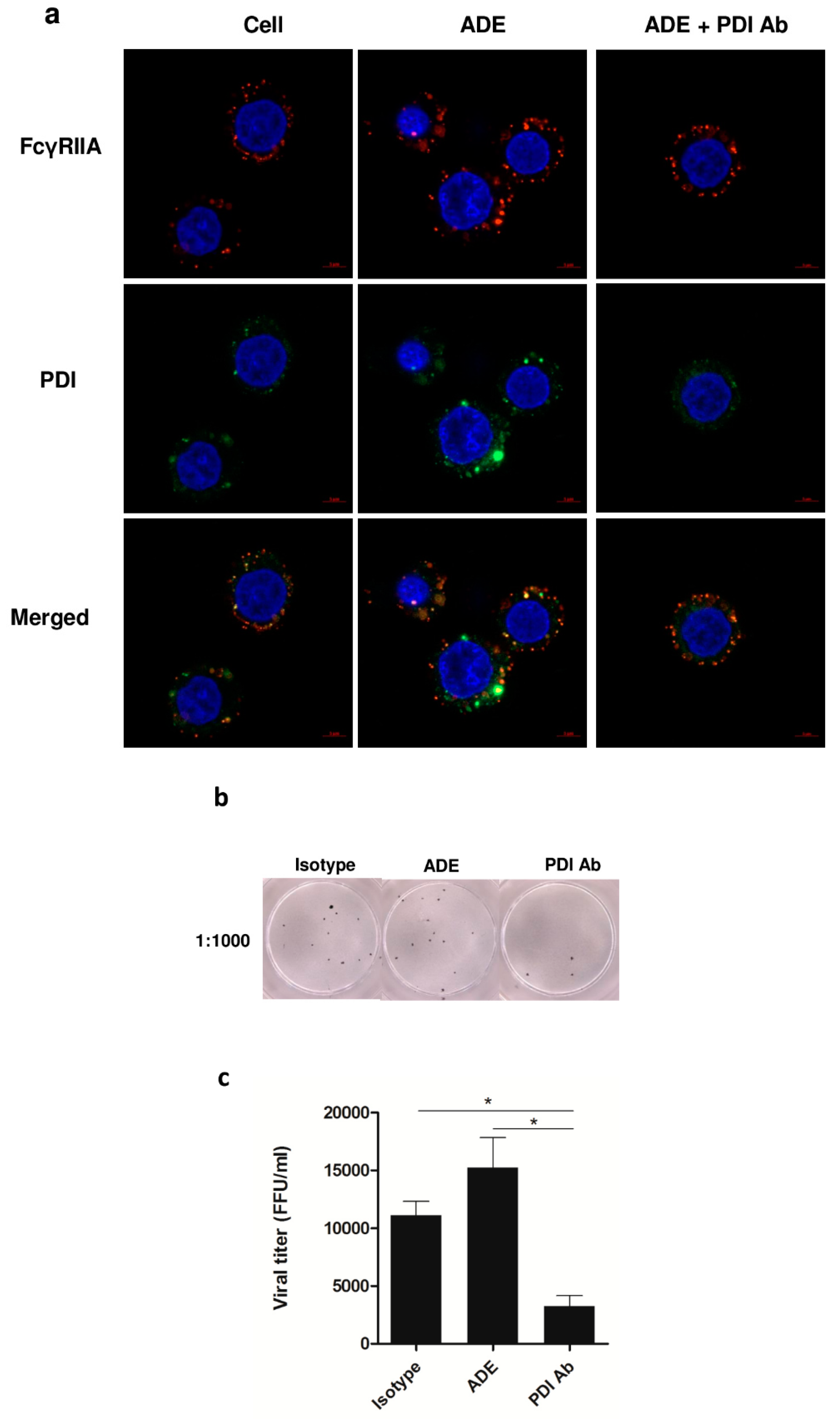
3.4. Role of PDI in DENV Infection, and Viral Protein Production during ADE Infection in U937 Cells
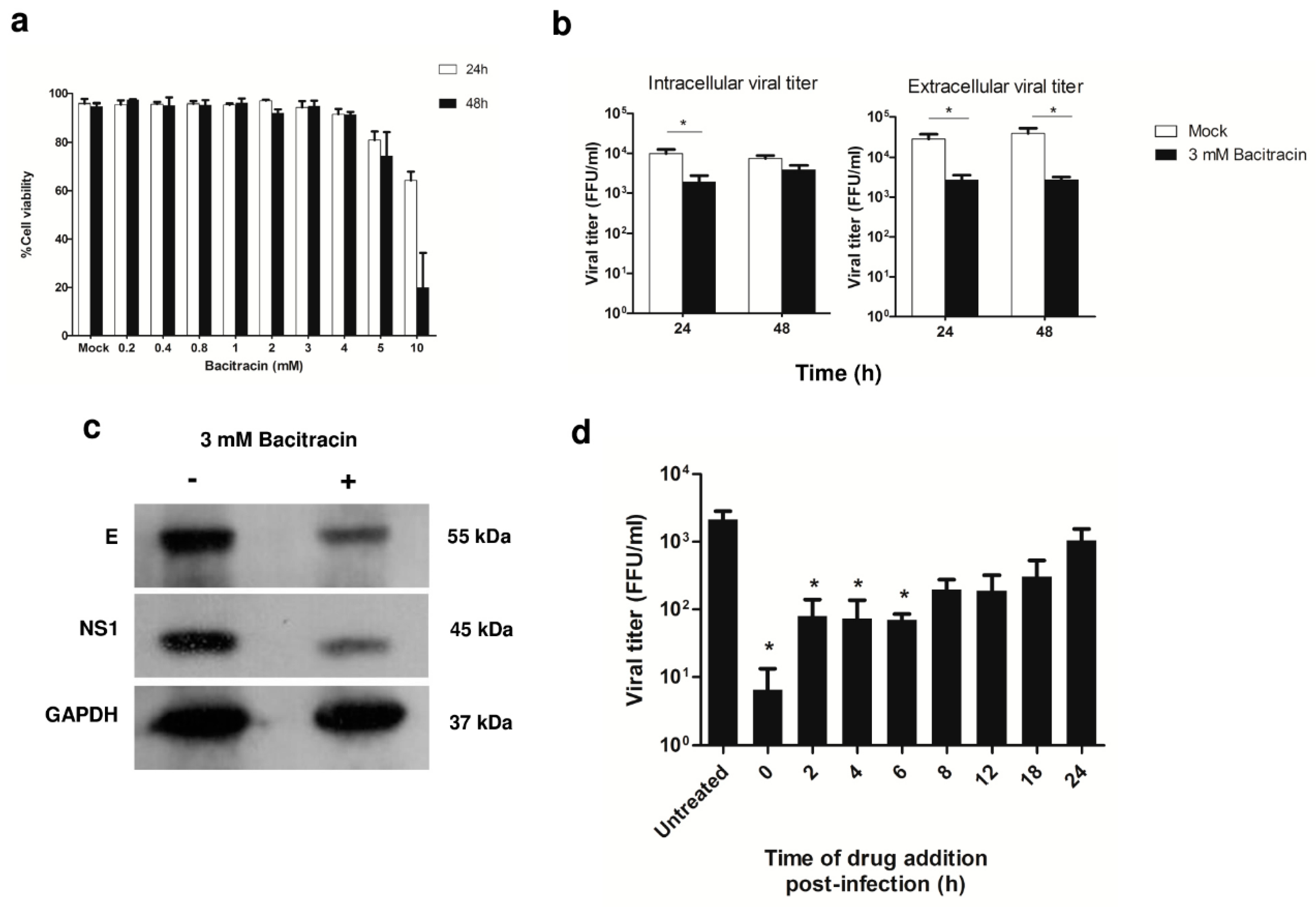
3.5. Effect of PDI Inhibition on Virus Life Cycle Stages
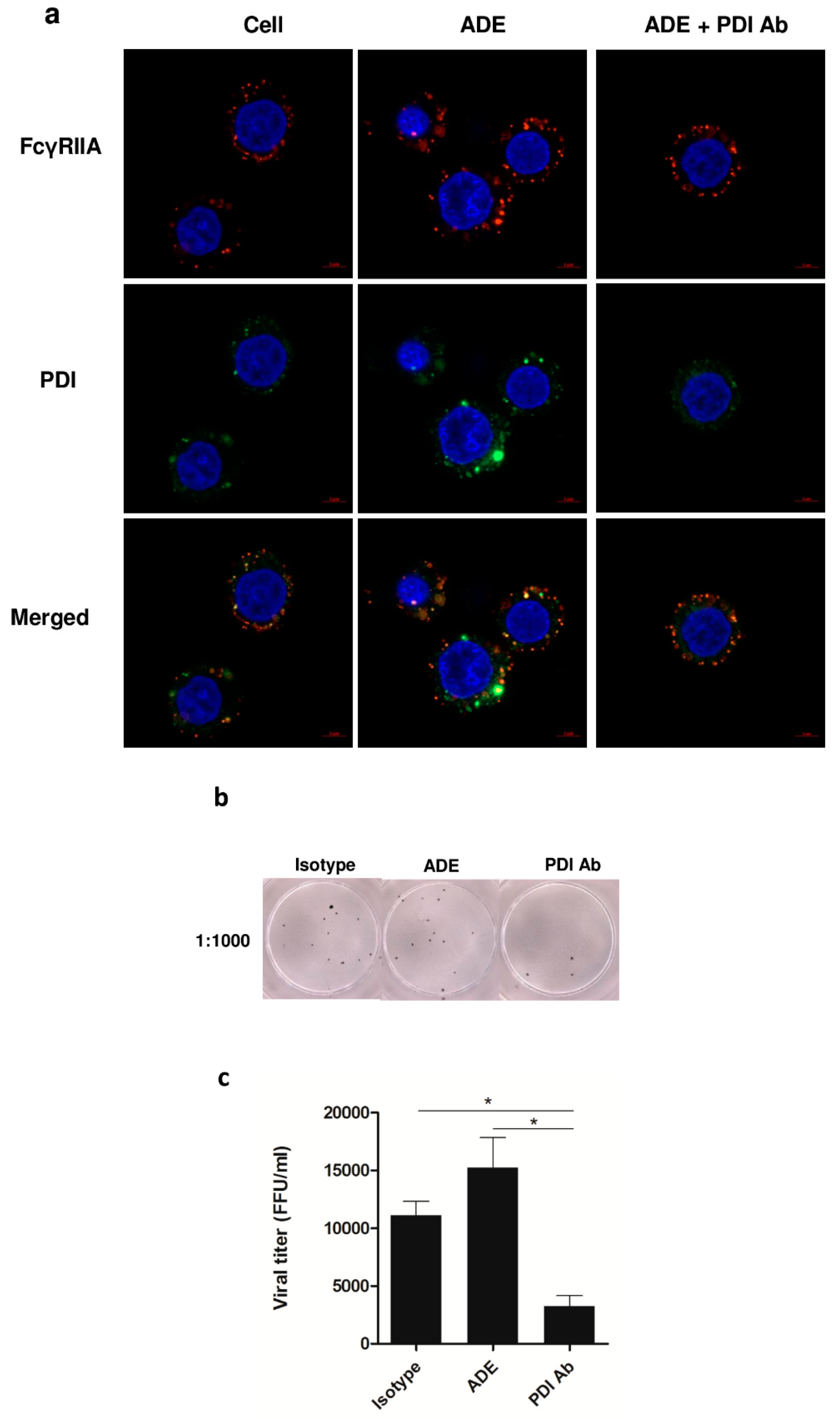
3.6. Significance of PDI in DENV Infection during ADE Infection
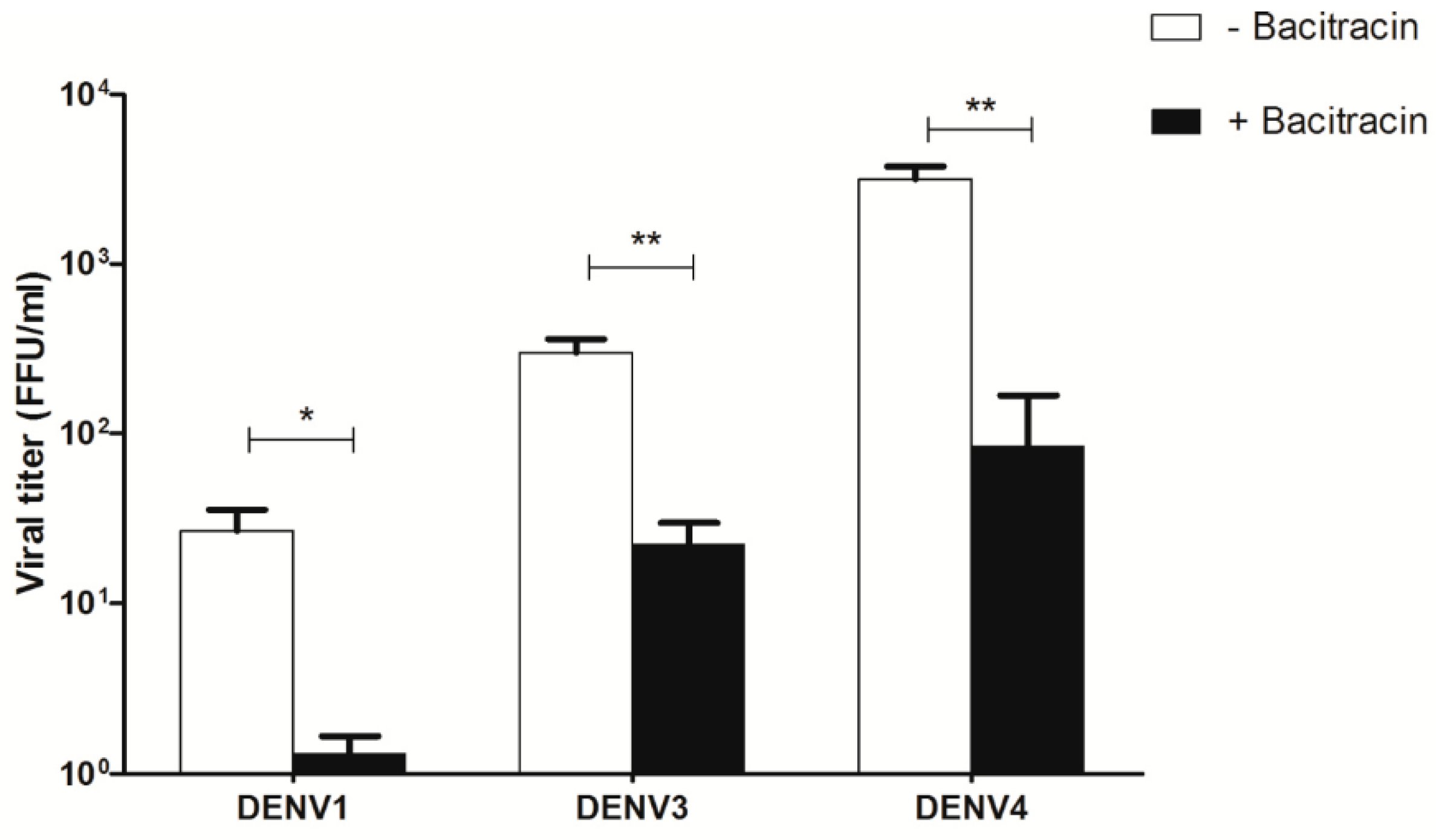
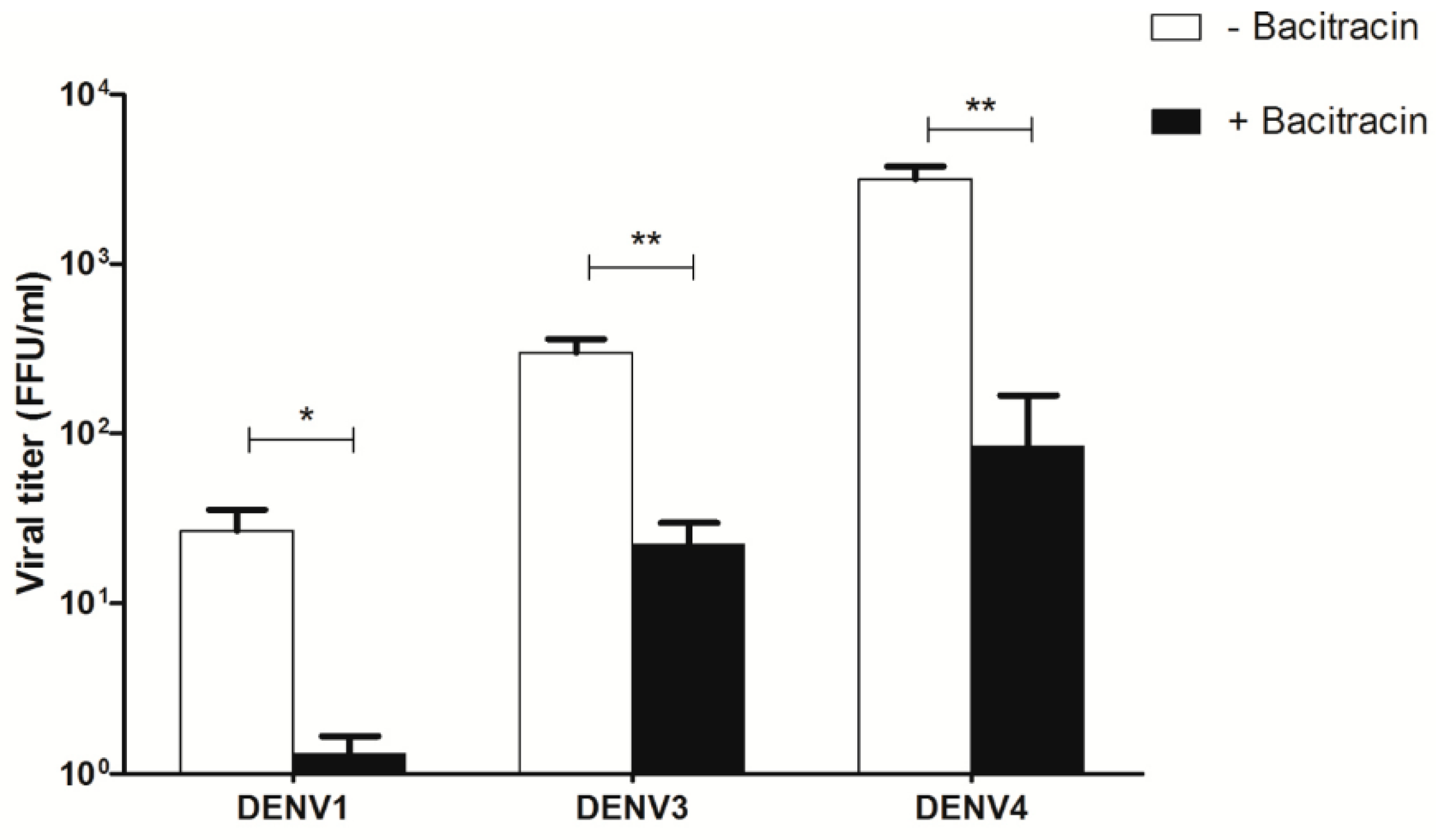
3.7. Effect of Bacitracin on Viral Titers of DENV1, DENV3, and DENV4 Infection of U937 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guha-Sapir, D.; Schimmer, B. Dengue fever: New paradigms for a changing epidemiology. Emerg. Themes Epidemiol. 2005, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Rodenhuis-Zybert, I.A.; Wilschut, J.; Smit, J.M. Dengue virus life cycle: Viral and host factors modulating infectivity. Cell. Mol. Life Sci. CMLS 2010, 67, 2773–2786. [Google Scholar] [CrossRef]
- Vasilakis, N.; Cardosa, J.; Hanley, K.A.; Holmes, E.C.; Weaver, S.C. Fever from the forest: Prospects for the continued emergence of sylvatic dengue virus and its impact on public health. Nat. Rev. Microbiol. 2011, 9, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Martina, B.E.; Koraka, P.; Osterhaus, A.D. Dengue virus pathogenesis: An integrated view. Clin. Microbiol. Rev. 2009, 22, 564–581. [Google Scholar] [CrossRef] [PubMed]
- Russell, P.K.; Udomsakdi, S.; Halstead, S.B. Antibody response in dengue and dengue hemorrhagic fever. Jpn. J. Med. Sci. biol. 1967, 20, 103–108. [Google Scholar]
- Kliks, S.C.; Nimmanitya, S.; Nisalak, A.; Burke, D.S. Evidence that maternal dengue antibodies are important in the development of dengue hemorrhagic fever in infants. Am. J. Trop. Med. Hyg. 1988, 38, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B.; O’Rourke, E.J. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J. Exp. Med. 1977, 146, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.G.; Kouri, G.; Bravo, J.; Valdes, L.; Vazquez, S.; Halstead, S.B. Effect of age on outcome of secondary dengue 2 infections. Int. J. Infect. Dis. 2002, 6, 118–124. [Google Scholar] [CrossRef]
- Katzelnick, L.C.; Gresh, L.; Halloran, M.E.; Mercado, J.C.; Kuan, G.; Gordon, A.; Balmaseda, A.; Harris, E. Antibody-dependent enhancement of severe dengue disease in humans. Science 2017, 358, 929–932. [Google Scholar] [CrossRef]
- Puerta-Guardo, H.; Mosso, C.; Medina, F.; Liprandi, F.; Ludert, J.E.; del Angel, R.M. Antibody-dependent enhancement of dengue virus infection in u937 cells requires cholesterol-rich membrane microdomains. J. Gen. Virol. 2010, 91, 394–403. [Google Scholar] [CrossRef]
- Goncalvez, A.P.; Engle, R.E.; St Claire, M.; Purcell, R.H.; Lai, C.J. Monoclonal antibody-mediated enhancement of dengue virus infection in vitro and in vivo and strategies for prevention. Proc. Natl. Acad. Sci. USA 2007, 104, 9422–9427. [Google Scholar] [CrossRef] [PubMed]
- Chareonsirisuthigul, T.; Kalayanarooj, S.; Ubol, S. Dengue virus (DENV) antibody-dependent enhancement of infection upregulates the production of anti-inflammatory cytokines, but suppresses anti-denv free radical and pro-inflammatory cytokine production, in thp-1 cells. J. Gen. Virol. 2007, 88, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M.; Halstead, S.B. Measurement of antibody-dependent infection enhancement of four dengue virus serotypes by monoclonal and polyclonal antibodies. J. Gen. Virol. 1990, 71 Pt 12, 2909–2914. [Google Scholar] [CrossRef]
- Huang, X.; Yue, Y.; Li, D.; Zhao, Y.; Qiu, L.; Chen, J.; Pan, Y.; Xi, J.; Wang, X.; Sun, Q.; et al. Antibody-dependent enhancement of dengue virus infection inhibits rlr-mediated type-i ifn-independent signalling through upregulation of cellular autophagy. Sci. Rep. 2016, 6, 22303. [Google Scholar] [CrossRef] [PubMed]
- Flipse, J.; Diosa-Toro, M.A.; Hoornweg, T.E.; van de Pol, D.P.; Urcuqui-Inchima, S.; Smit, J.M. Antibody-dependent enhancement of dengue virus infection in primary human macrophages; balancing higher fusion against antiviral responses. Sci. Rep. 2016, 6, 29201. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.T.; Chuang, Y.J.; Lin, Y.S.; Chang, C.P.; Wan, S.W.; Lin, S.H.; Chen, C.L.; Lin, C.F. Antibody-dependent enhancement infection facilitates dengue virus-regulated signaling of IL-10 production in monocytes. PLoS Negl. Trop. Dis. 2014, 8, e3320. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, W.; Marzi, A.; Carmody, A.B.; Maruyama, J.; Kuroda, M.; Miyamoto, H.; Nanbo, A.; Manzoor, R.; Yoshida, R.; Igarashi, M.; et al. Fcγ-receptor iia-mediated src signaling pathway is essential for the antibody-dependent enhancement of ebola virus infection. PLoS Pathog. 2016, 12, e1006139. [Google Scholar] [CrossRef]
- Shresta, S.; Kyle, J.L.; Snider, H.M.; Basavapatna, M.; Beatty, P.R.; Harris, E. Interferon-dependent immunity is essential for resistance to primary dengue virus infection in mice, whereas t- and b-cell-dependent immunity are less critical. J. Virol. 2004, 78, 2701–2710. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.G.; Robinson, P.J.; Bletchly, C.; Mackenzie, J.M.; Young, P.R. Dengue virus nonstructural protein 1 is expressed in a glycosyl-phosphatidylinositol-linked form that is capable of signal transduction. FASEB J. 2000, 14, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Image Processing and Analysis in Java. 2 October 2012. Available online: https://imagej.nih.gov/ij/download.html (accessed on 27 January 2019).
- Kou, Z.; Quinn, M.; Chen, H.; Rodrigo, W.W.; Rose, R.C.; Schlesinger, J.J.; Jin, X. Monocytes, but not t or b cells, are the principal target cells for dengue virus (dv) infection among human peripheral blood mononuclear cells. J. Med. Virol. 2008, 80, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Jessie, K.; Fong, M.Y.; Devi, S.; Lam, S.K.; Wong, K.T. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J. Infect. Dis. 2004, 189, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Watts, D.M.; Porter, K.R.; Putvatana, P.; Vasquez, B.; Calampa, C.; Hayes, C.G.; Halstead, S.B. Failure of secondary infection with american genotype dengue 2 to cause dengue haemorrhagic fever. Lancet 1999, 354, 1431–1434. [Google Scholar] [CrossRef]
- Moi, M.L.; Lim, C.K.; Kotaki, A.; Takasaki, T.; Kurane, I. Development of an antibody-dependent enhancement assay for dengue virus using stable bhk-21 cell lines expressing fc gammariia. J. Virol. Methods 2010, 163, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zou, L.; Hu, Z.; Chen, W.; Zhang, J.; Zhu, J.; Fang, X.; Yuan, W.; Hu, X.; Hu, F.; et al. Identification and characterization of a 43 kda actin protein involved in the denv-2 binding and infection of ecv304 cells. Microbes Infect. Inst. Pasteur 2013, 15, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Zhang, J.L.; Chen, W.; Xu, X.F.; Gao, N.; Fan, D.Y.; An, J. Roles of small gtpase rac1 in the regulation of actin cytoskeleton during dengue virus infection. PLoS Negl. Trop. Dis. 2010, 4, e809. [Google Scholar] [CrossRef] [PubMed]
- Kanlaya, R.; Pattanakitsakul, S.N.; Sinchaikul, S.; Chen, S.T.; Thongboonkerd, V. Vimentin interacts with heterogeneous nuclear ribonucleoproteins and dengue nonstructural protein 1 and is important for viral replication and release. Mol. Biosyst. 2010, 6, 795–806. [Google Scholar] [CrossRef]
- Chiou, C.T.; Hu, C.C.; Chen, P.H.; Liao, C.L.; Lin, Y.L.; Wang, J.J. Association of japanese encephalitis virus ns3 protein with microtubules and tumour susceptibility gene 101 (tsg101) protein. J. Gen. Virol. 2003, 84, 2795–2805. [Google Scholar] [CrossRef]
- Turano, C.; Coppari, S.; Altieri, F.; Ferraro, A. Proteins of the pdi family: Unpredicted non-er locations and functions. J. Cell. Physiol. 2002, 193, 154–163. [Google Scholar] [CrossRef]
- Ou, W.; Silver, J. Role of protein disulfide isomerase and other thiol-reactive proteins in hiv-1 envelope protein-mediated fusion. Virology 2006, 350, 406–417. [Google Scholar] [CrossRef]
- Auwerx, J.; Isacsson, O.; Soderlund, J.; Balzarini, J.; Johansson, M.; Lundberg, M. Human glutaredoxin-1 catalyzes the reduction of HIV-1 gp120 and cd4 disulfides and its inhibition reduces hiv-1 replication. Int. J. Biochem. Cell Biol. 2009, 41, 1269–1275. [Google Scholar] [CrossRef]
- Gilbert, J.; Ou, W.; Silver, J.; Benjamin, T. Downregulation of protein disulfide isomerase inhibits infection by the mouse polyomavirus. J. Virol. 2006, 80, 10868–10870. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; McGinnes, L.W.; Morrison, T.G. Role of thiol/disulfide exchange in newcastle disease virus entry. J. Virol. 2009, 83, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.W.; Lin, C.F.; Lu, Y.T.; Lei, H.Y.; Anderson, R.; Lin, Y.S. Endothelial cell surface expression of protein disulfide isomerase activates beta1 and beta3 integrins and facilitates dengue virus infection. J. Cell. Biochem. 2012, 113, 1681–1691. [Google Scholar] [PubMed]
- Zhang, J.L.; Wang, J.L.; Gao, N.; Chen, Z.T.; Tian, Y.P.; An, J. Up-regulated expression of beta3 integrin induced by dengue virus serotype 2 infection associated with virus entry into human dermal microvascular endothelial cells. Biochem. Biophys. Res. Commun. 2007, 356, 763–768. [Google Scholar] [CrossRef]
- Diwaker, D.; Mishra, K.P.; Ganju, L.; Singh, S.B. Protein disulfide isomerase mediates dengue virus entry in association with lipid rafts. Viral Immunol. 2015, 28, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.A. Bacitracin: An inhibitor of the insulin degrading activity of glutathione-insulin transhydrogenase. Biochem. Biophys. Res. Commun. 1981, 98, 431–438. [Google Scholar] [CrossRef]
- Dickerhof, N.; Kleffmann, T.; Jack, R.; McCormick, S. Bacitracin inhibits the reductive activity of protein disulfide isomerase by disulfide bond formation with free cysteines in the substrate-binding domain. FEBS J. 2011, 278, 2034–2043. [Google Scholar] [CrossRef]
- Zhou, M.; Jacob, A.; Ho, N.; Miksa, M.; Wu, R.; Maitra, S.R.; Wang, P. Downregulation of protein disulfide isomerase in sepsis and its role in tumor necrosis factor-alpha release. Crit. Care 2008, 12, R100. [Google Scholar] [CrossRef]
- Wati, S.; Soo, M.L.; Zilm, P.; Li, P.; Paton, A.W.; Burrell, C.J.; Beard, M.; Carr, J.M. Dengue virus infection induces upregulation of grp78, which acts to chaperone viral antigen production. J. Virol. 2009, 83, 12871–12880. [Google Scholar] [CrossRef]
- Ko, H.S.; Uehara, T.; Nomura, Y. Role of ubiquilin associated with protein-disulfide isomerase in the endoplasmic reticulum in stress-induced apoptotic cell death. J. Biol. Chem. 2002, 277, 35386–35392. [Google Scholar] [CrossRef]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef] [PubMed]
- Qing, M.; Yang, F.; Zhang, B.; Zou, G.; Robida, J.M.; Yuan, Z.; Tang, H.; Shi, P.Y. Cyclosporine inhibits flavivirus replication through blocking the interaction between host cyclophilins and viral ns5 protein. Antimicrob. Agents Chemother. 2009, 53, 3226–3235. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.Y.; Kondreddi, R.R.; Xie, X.; Rao, R.; Nilar, S.; Xu, H.Y.; Qing, M.; Chang, D.; Dong, H.; Yokokawa, F.; et al. A translation inhibitor that suppresses dengue virus in vitro and in vivo. Antimicrob. Agents Chemother. 2011, 55, 4072–4080. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, E.; Brown, E.J.; Rosales, C. Transmembrane mutations to fcgammariia alter its association with lipid rafts: Implications for receptor signaling. J. Immunol. 2007, 178, 3048–3058. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, K.; Sobota, A. The clustered fcgamma receptor ii is recruited to lyn-containing membrane domains and undergoes phosphorylation in a cholesterol-dependent manner. Eur. J. Immunol. 2001, 31, 989–998. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rawarak, N.; Suttitheptumrong, A.; Reamtong, O.; Boonnak, K.; Pattanakitsakul, S.-n. Protein Disulfide Isomerase Inhibitor Suppresses Viral Replication and Production during Antibody-Dependent Enhancement of Dengue Virus Infection in Human Monocytic Cells. Viruses 2019, 11, 155. https://doi.org/10.3390/v11020155
Rawarak N, Suttitheptumrong A, Reamtong O, Boonnak K, Pattanakitsakul S-n. Protein Disulfide Isomerase Inhibitor Suppresses Viral Replication and Production during Antibody-Dependent Enhancement of Dengue Virus Infection in Human Monocytic Cells. Viruses. 2019; 11(2):155. https://doi.org/10.3390/v11020155
Chicago/Turabian StyleRawarak, Nantapon, Aroonroong Suttitheptumrong, Onrapak Reamtong, Kobporn Boonnak, and Sa-nga Pattanakitsakul. 2019. "Protein Disulfide Isomerase Inhibitor Suppresses Viral Replication and Production during Antibody-Dependent Enhancement of Dengue Virus Infection in Human Monocytic Cells" Viruses 11, no. 2: 155. https://doi.org/10.3390/v11020155
APA StyleRawarak, N., Suttitheptumrong, A., Reamtong, O., Boonnak, K., & Pattanakitsakul, S.-n. (2019). Protein Disulfide Isomerase Inhibitor Suppresses Viral Replication and Production during Antibody-Dependent Enhancement of Dengue Virus Infection in Human Monocytic Cells. Viruses, 11(2), 155. https://doi.org/10.3390/v11020155