Divergent Evolution of E1A CR3 in Human Adenovirus Species D

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Analysis
2.2. Proteotyping
2.3. Protein Sequence and Structural Analysis
2.4. Codon Usage Bias Analysis
3. Results
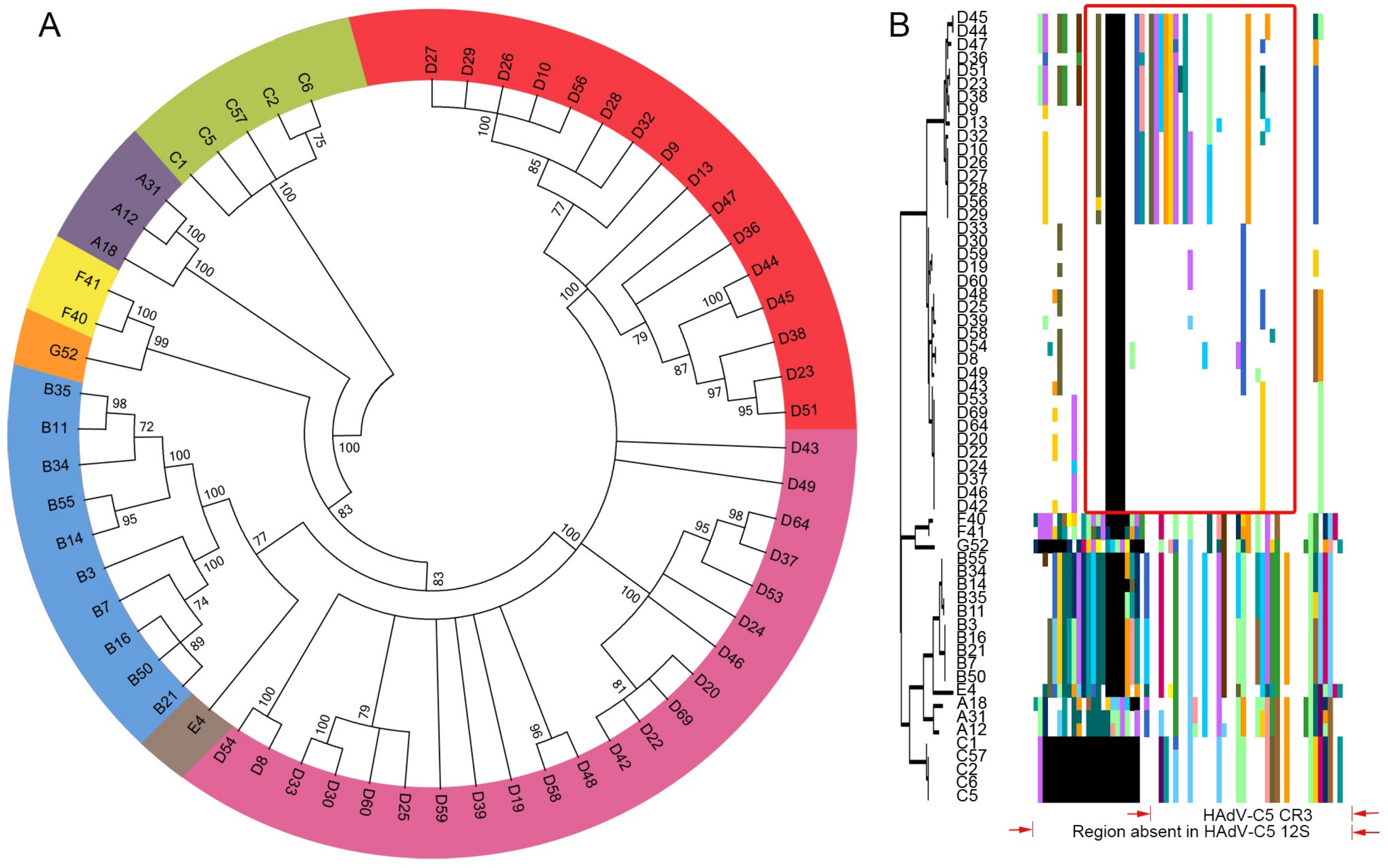
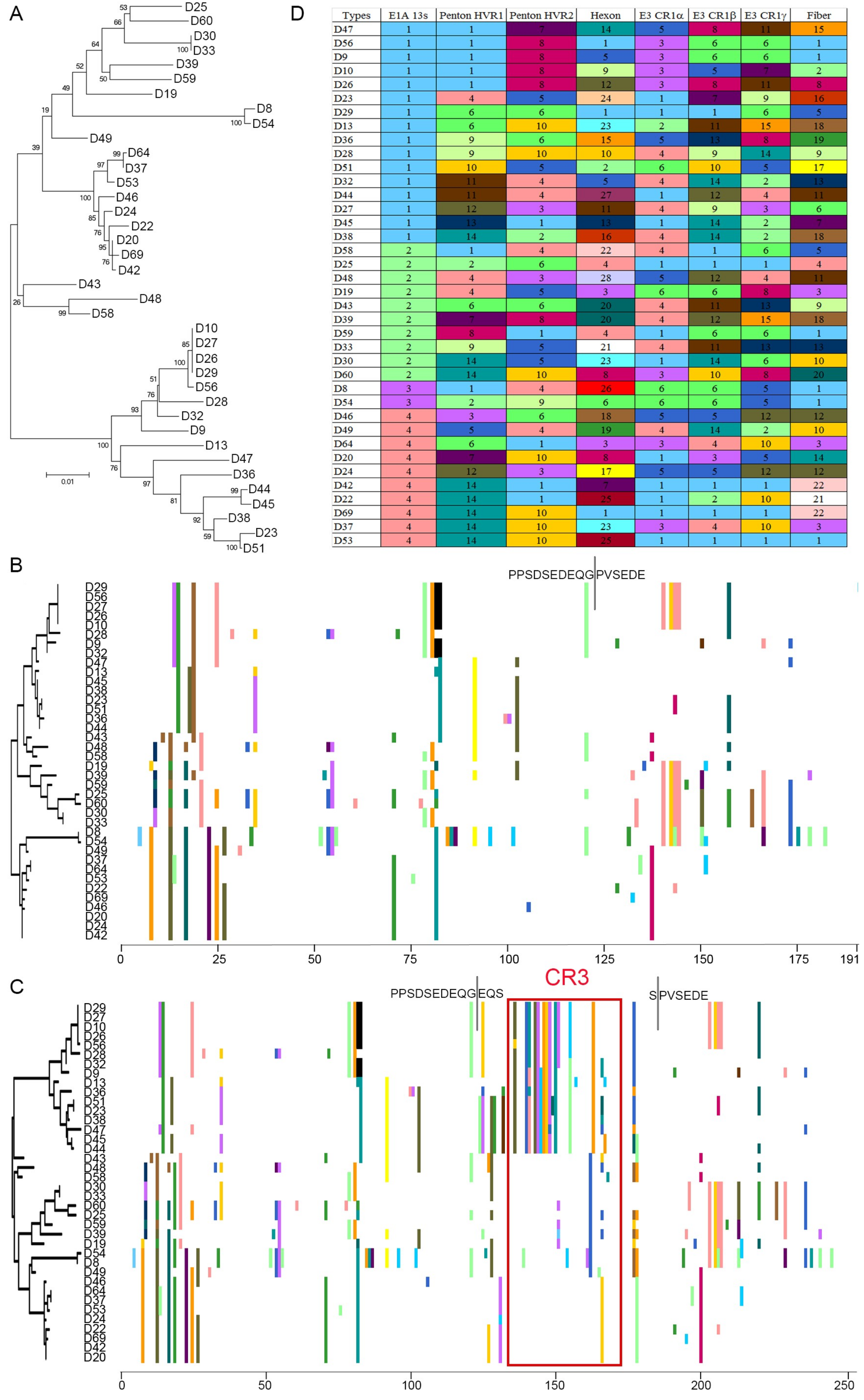
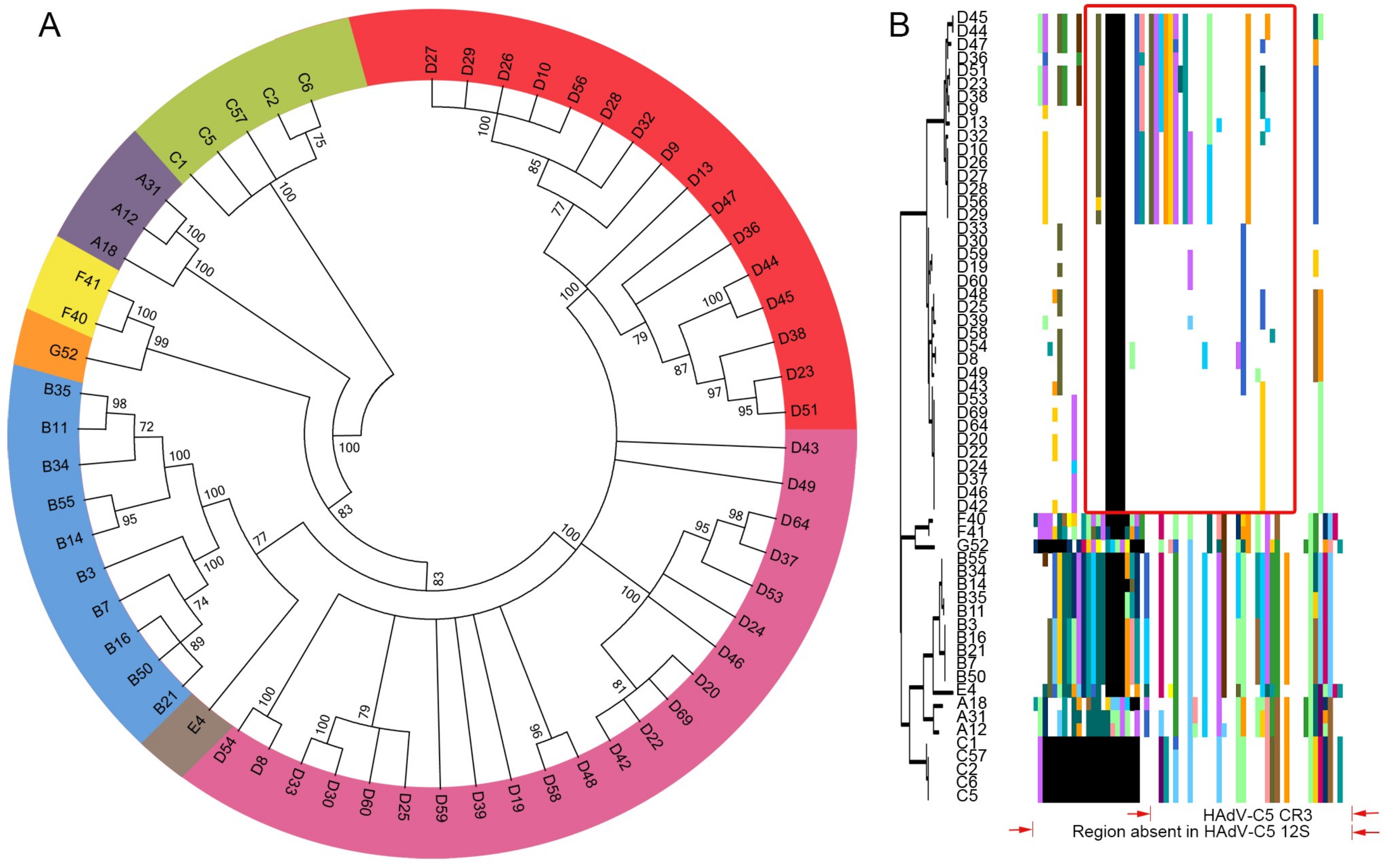
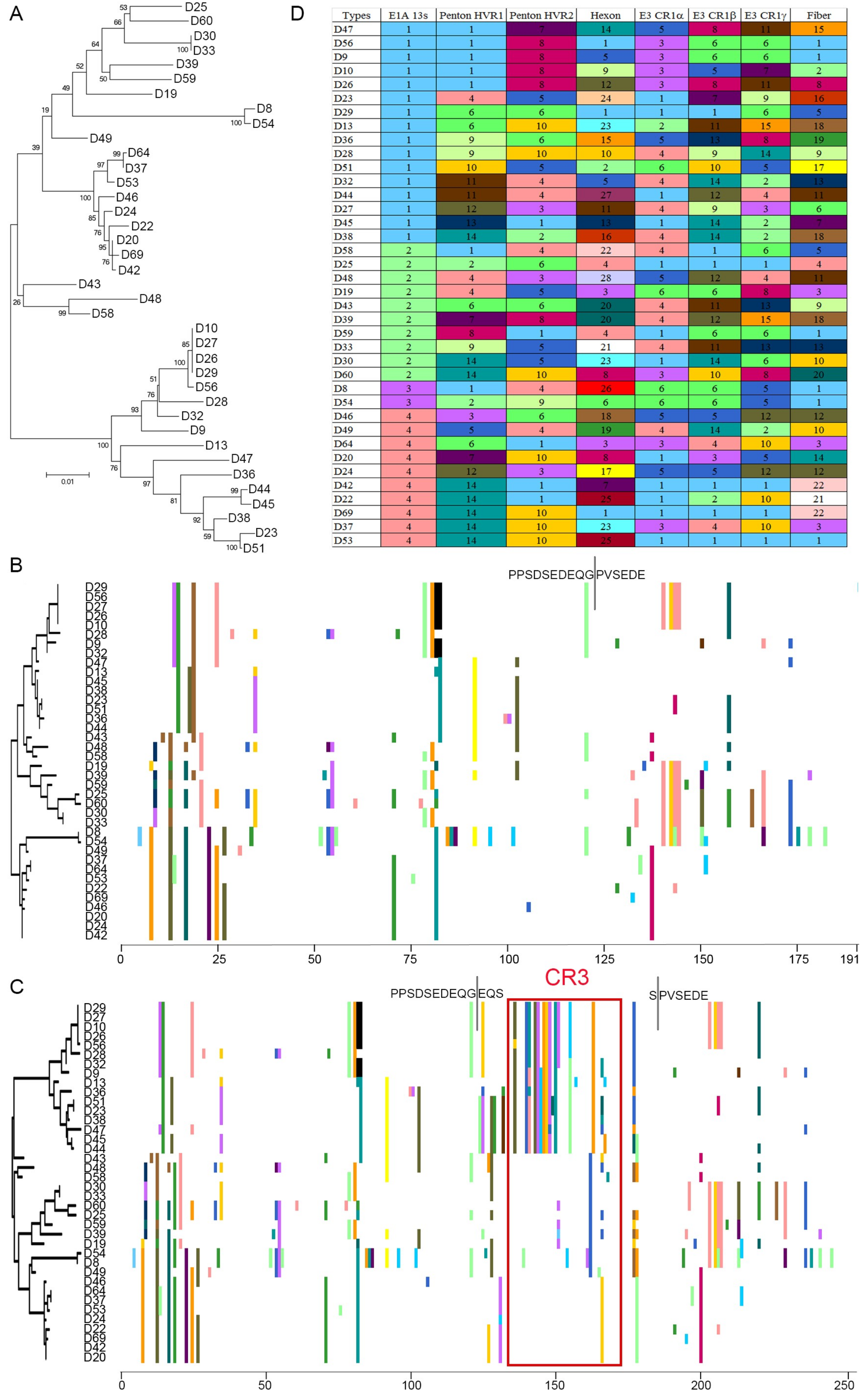
3.1. Segregation of the 13S Isoform of HAdV-D E1A into Two Major Subclades
3.2. Evidence for Prior Homologous Recombination in HAdV-D E1A
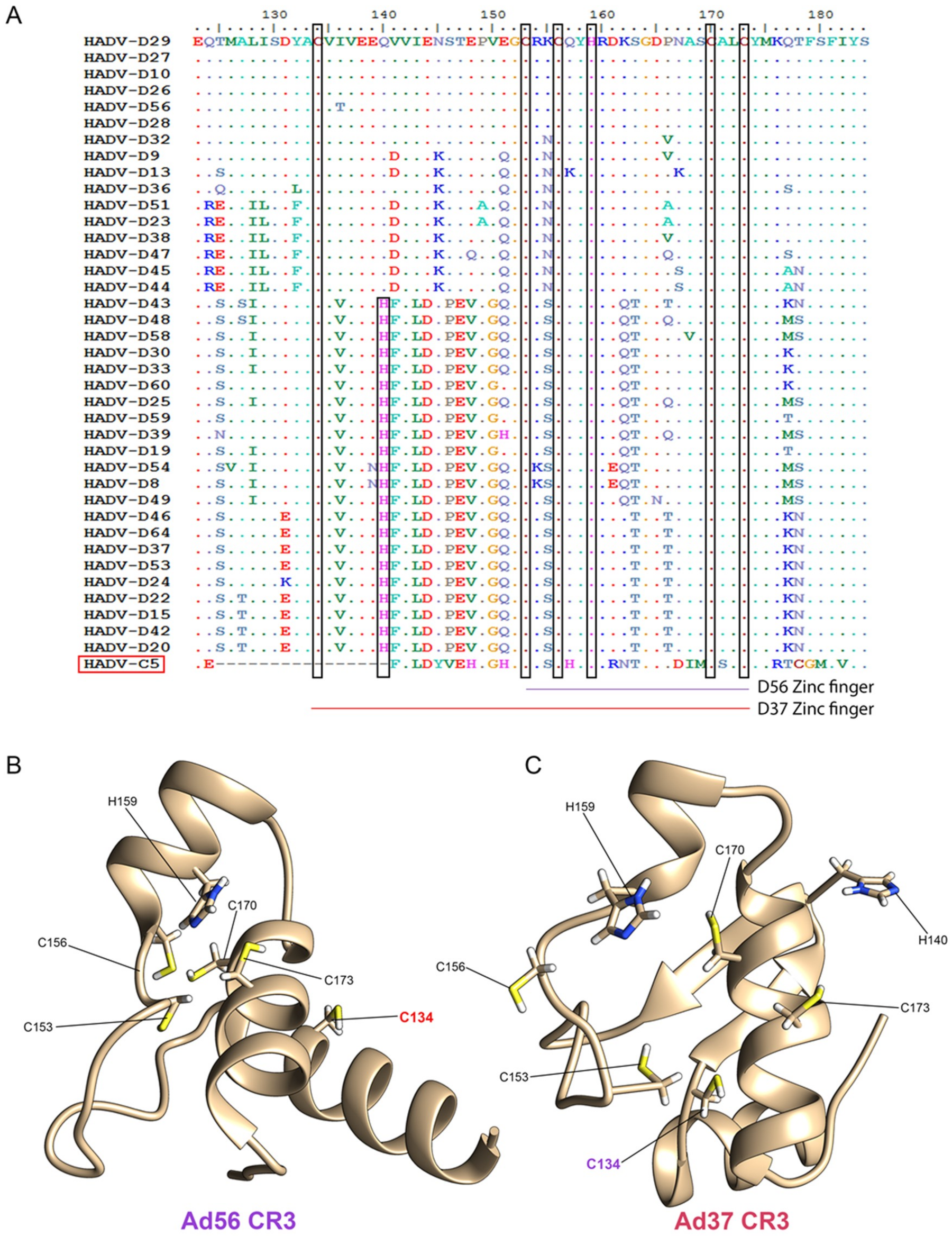
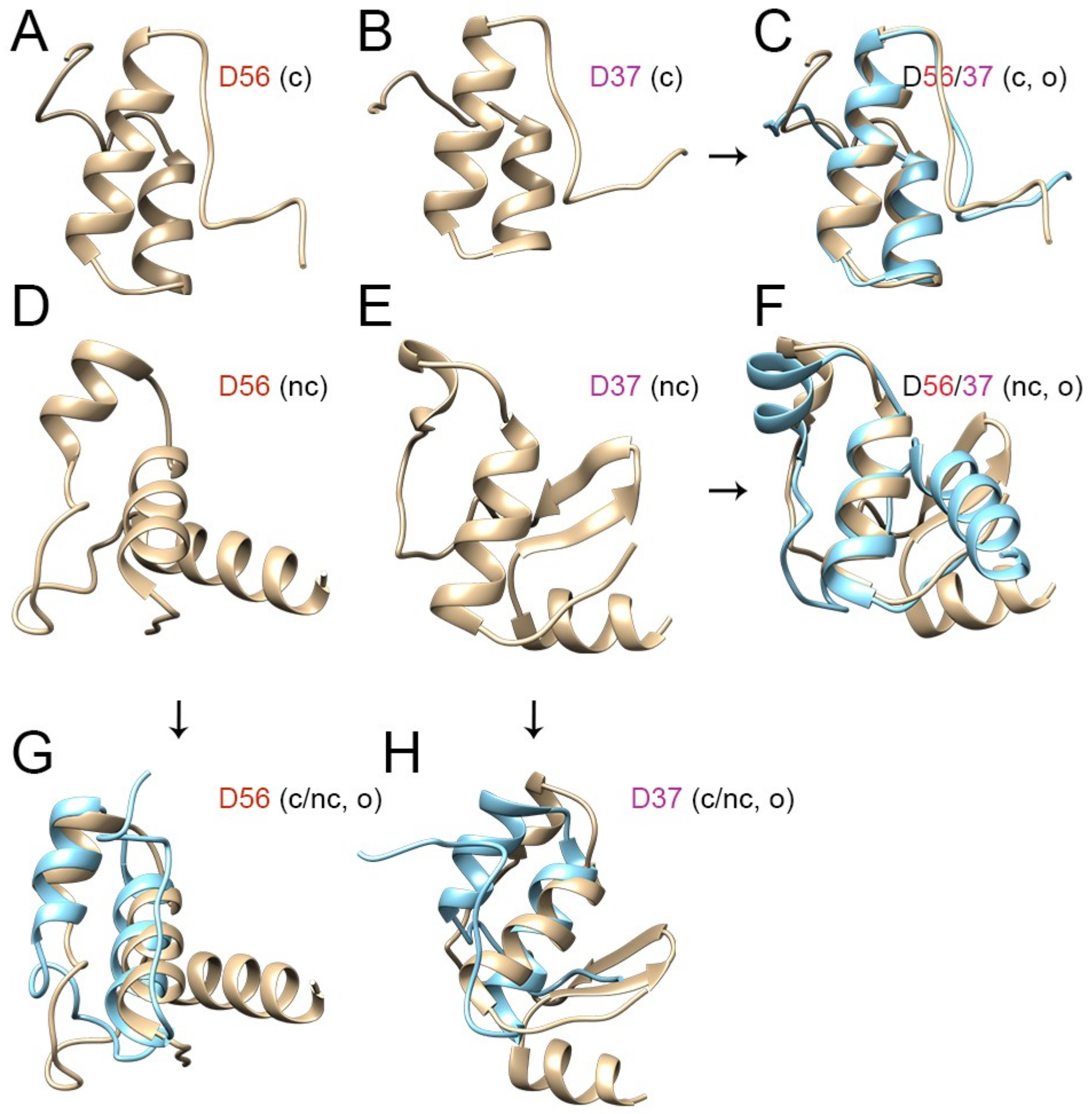
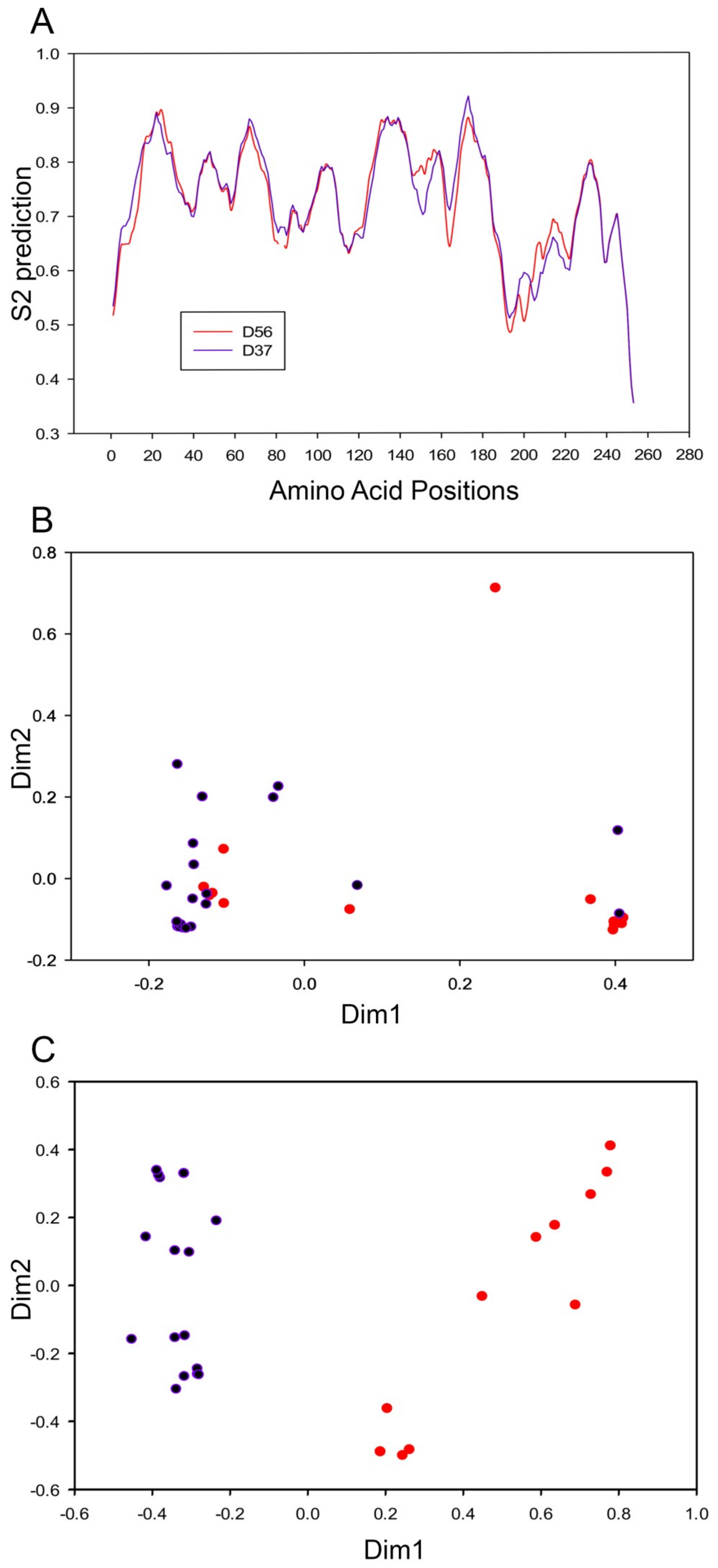
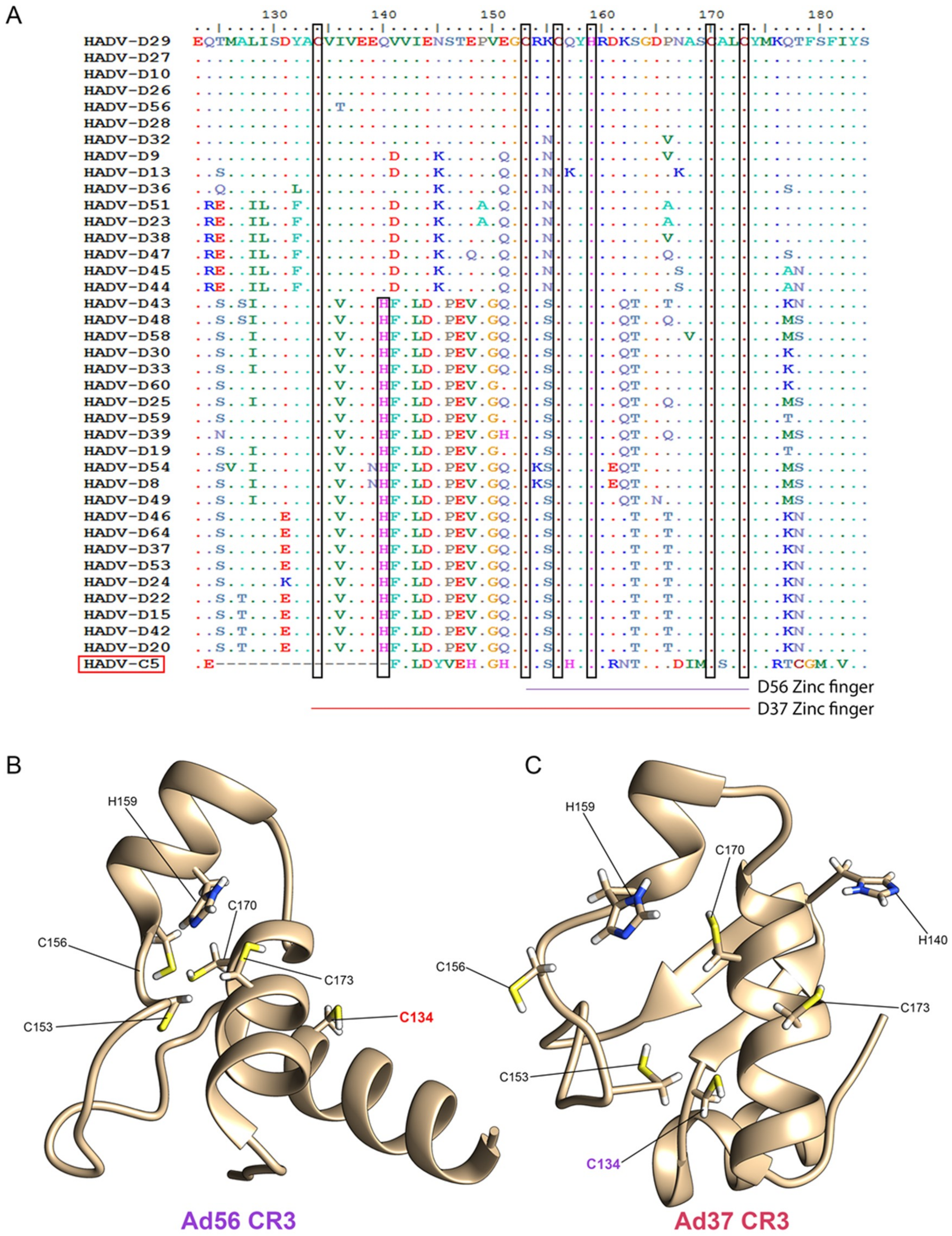
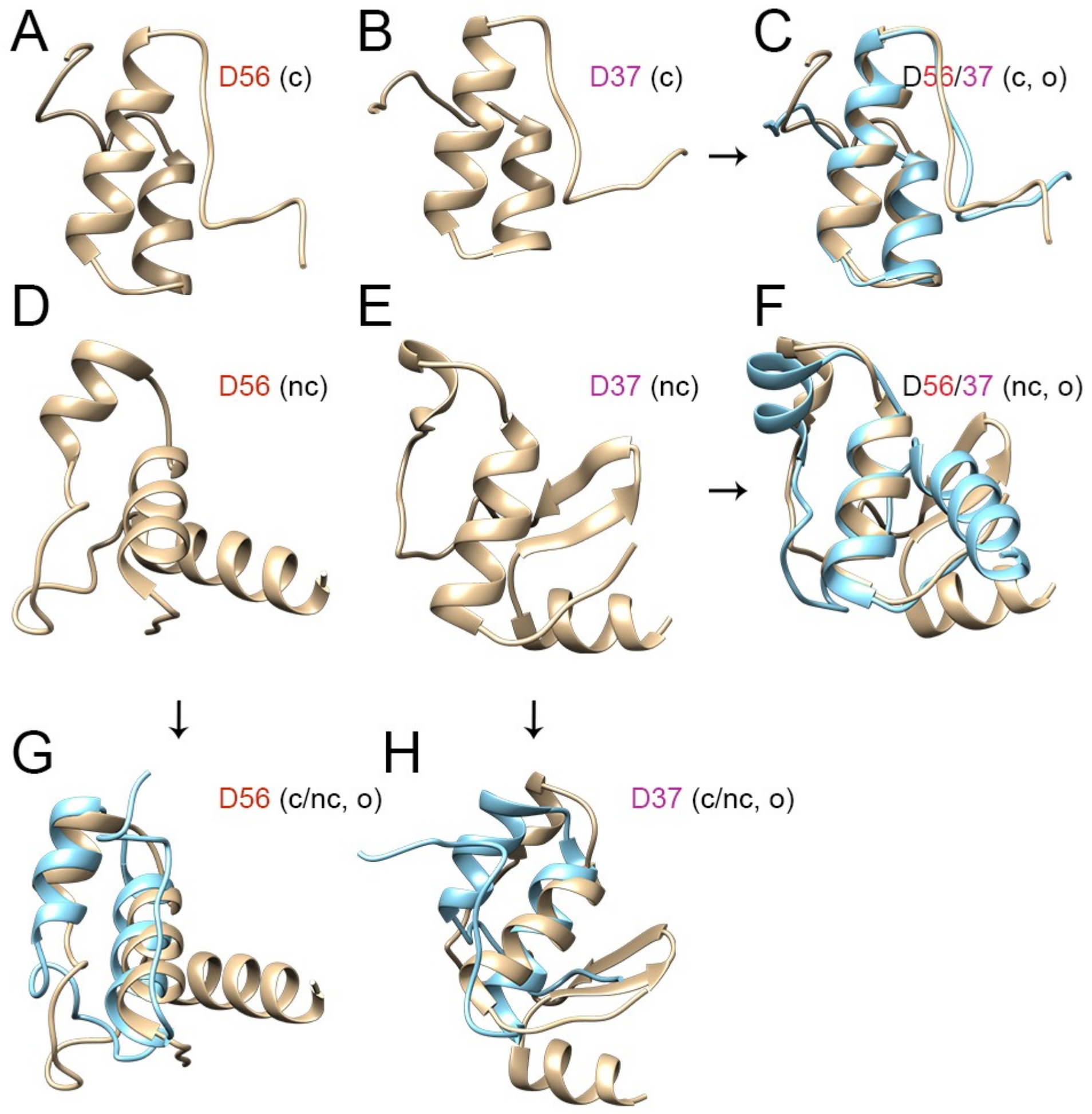
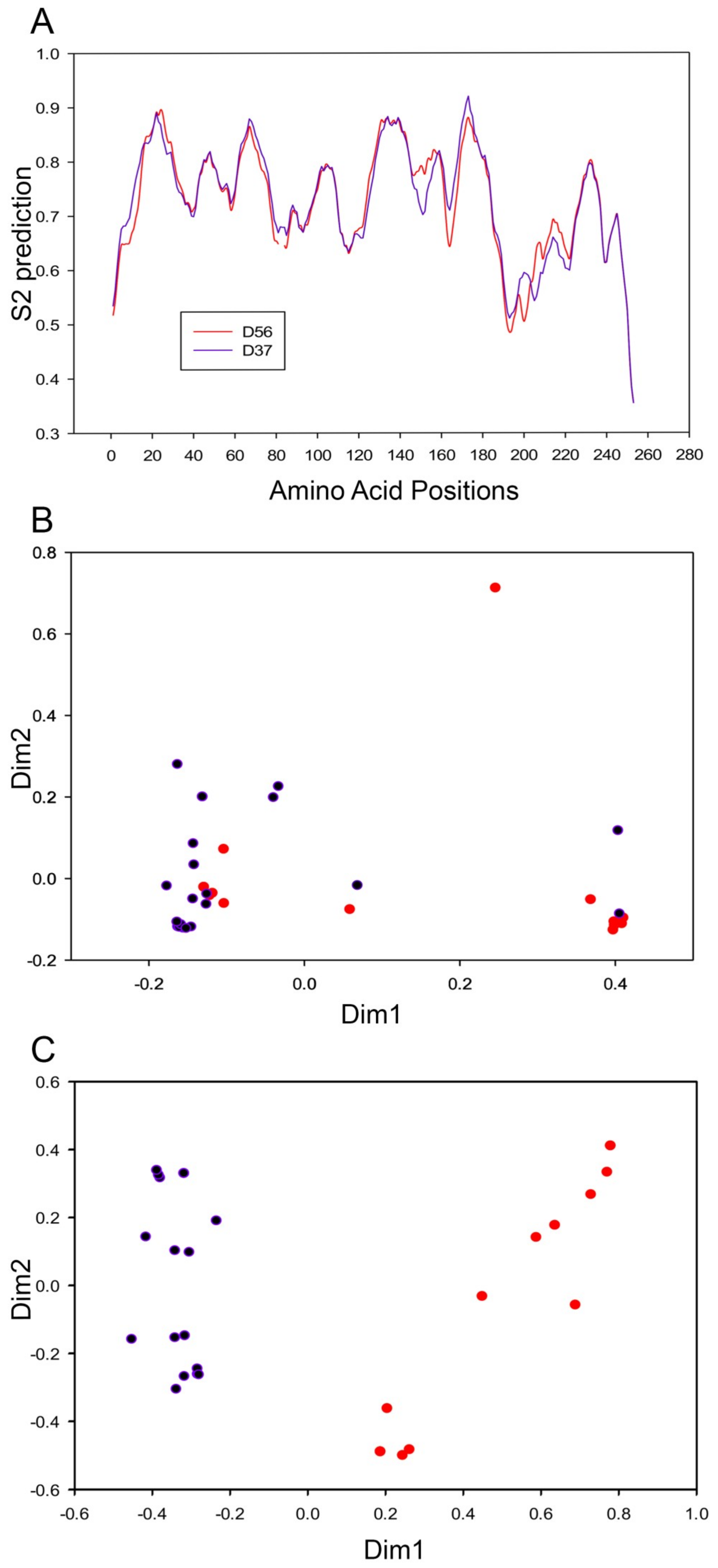
3.3. Evolutionary Divergence in HAdV-D E1A 13S Zinc Finger-Containing CR3 Region
3.4. Codon Usage Bias Analysis and Divergent Evolution of HAdV-D E1A 13S
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Robinson, C.M.; Singh, G.; Henquell, C.; Walsh, M.P.; Peigue-Lafeuille, H.; Seto, D.; Jones, M.S.; Dyer, D.W.; Chodosh, J. Computational analysis and identification of an emergent human adenovirus pathogen implicated in a respiratory fatality. Virology 2011, 409, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Robinson, C.M.; Dehghan, S.; Schmidt, T.; Seto, D.; Jones, M.S.; Dyer, D.W.; Chodosh, J. Overreliance on the hexon gene, leading to misclassification of human adenoviruses. J. Virol. 2012, 86, 4693–4695. [Google Scholar] [CrossRef]
- Robinson, C.M.; Zhou, X.; Rajaiya, J.; Yousuf, M.A.; Singh, G.; DeSerres, J.J.; Walsh, M.P.; Wong, S.; Seto, D.; Dyer, D.W.; et al. Predicting the next eye pathogen: Analysis of a novel adenovirus. mBio 2013, 4, e00595-12. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Zhou, X.; Lee, J.Y.; Yousuf, M.A.; Ramke, M.; Ismail, A.M.; Lee, J.S.; Robinson, C.M.; Seto, D.; Dyer, D.W.; et al. Recombination of the epsilon determinant and corneal tropism: Human adenovirus species D types 15, 29, 56, and 69. Virology 2015, 485, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Singh, G.; Lee, J.Y.; Dehghan, S.; Rajaiya, J.; Liu, E.B.; Yousuf, M.A.; Betensky, R.A.; Jones, M.S.; Dyer, D.W.; et al. Molecular evolution of human adenoviruses. Sci. Rep. 2013, 3, 1812. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Robinson, C.M.; Dehghan, S.; Jones, M.S.; Dyer, D.W.; Seto, D.; Chodosh, J. Homologous recombination in E3 genes of human adenovirus species D. J. Virol. 2013, 87, 12481–12488. [Google Scholar] [CrossRef] [PubMed]
- Dehghan, S.; Seto, J.; Liu, E.B.; Walsh, M.P.; Dyer, D.W.; Chodosh, J.; Seto, D. Computational analysis of four human adenovirus type 4 genomes reveals molecular evolution through two interspecies recombination events. Virology 2013, 443, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.P.; Chintakuntlawar, A.; Robinson, C.M.; Madisch, I.; Harrach, B.; Hudson, N.R.; Schnurr, D.; Heim, A.; Chodosh, J.; Seto, D.; et al. Evidence of molecular evolution driven by recombination events influencing tropism in a novel human adenovirus that causes epidemic keratoconjunctivitis. PLoS ONE 2009, 4, e5635. [Google Scholar] [CrossRef]
- Walsh, M.P.; Seto, J.; Jones, M.S.; Chodosh, J.; Xu, W.; Seto, D. Computational analysis identifies human adenovirus type 55 as a re-emergent acute respiratory disease pathogen. J. Clin. Microbiol. 2010, 48, 991–993. [Google Scholar] [CrossRef]
- Seto, D.; Chodosh, J.; Brister, J.R.; Jones, M.S.; Members of the Adenovirus Research, C. Using the whole-genome sequence to characterize and name human adenoviruses. J. Virol. 2011, 85, 5701–5702. [Google Scholar] [CrossRef]
- Nevins, J.R.; Ginsberg, H.S.; Blanchard, J.M.; Wilson, M.C.; Darnell, J.E., Jr. Regulation of the primary expression of the early adenovirus transcription units. J. Virol. 1979, 32, 727–733. [Google Scholar] [PubMed]
- White, E. Regulation of the cell cycle and apoptosis by the oncogenes of adenovirus. Oncogene 2001, 20, 7836–7846. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Mymryk, J.S. Adenovirus-5 E1A: Paradox and paradigm. Nat. Rev. Mol. Cell Biol. 2002, 3, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Perricaudet, M.; Akusjarvi, G.; Virtanen, A.; Pettersson, U. Structure of two spliced mRNAs from the transforming region of human subgroup C adenoviruses. Nature 1979, 281, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Kimelman, D.; Miller, J.S.; Porter, D.; Roberts, B.E. E1a regions of the human adenoviruses and of the highly oncogenic simian adenovirus 7 are closely related. J. Virol. 1985, 53, 399–409. [Google Scholar] [PubMed]
- Avvakumov, N.; Wheeler, R.; D’Halluin, J.C.; Mymryk, J.S. Comparative sequence analysis of the largest E1A proteins of human and simian adenoviruses. J. Virol. 2002, 76, 7968–7975. [Google Scholar] [CrossRef]
- Avvakumov, N.; Kajon, A.E.; Hoeben, R.C.; Mymryk, J.S. Comprehensive sequence analysis of the E1A proteins of human and simian adenoviruses. Virology 2004, 329, 477–492. [Google Scholar] [CrossRef]
- Berk, A.J.; Lee, F.; Harrison, T.; Williams, J.; Sharp, P.A. Pre-early adenovirus 5 gene product regulates synthesis of early viral messenger RNAs. Cell 1979, 17, 935–944. [Google Scholar] [CrossRef]
- Jones, N.; Shenk, T. An adenovirus type 5 early gene function regulates expression of other early viral genes. Proc. Natl. Acad. Sci. USA 1979, 76, 3665–3669. [Google Scholar] [CrossRef]
- Ablack, J.N.; Pelka, P.; Yousef, A.F.; Turnell, A.S.; Grand, R.J.; Mymryk, J.S. Comparison of E1A CR3-dependent transcriptional activation across six different human adenovirus subgroups. J. Virol. 2010, 84, 12771–12781. [Google Scholar] [CrossRef]
- Boyer, T.G.; Berk, A.J. Functional interaction of adenovirus E1A with holo-TFIID. Genes Dev. 1993, 7, 1810–1823. [Google Scholar] [CrossRef] [PubMed]
- Geisberg, J.V.; Lee, W.S.; Berk, A.J.; Ricciardi, R.P. The zinc finger region of the adenovirus E1A transactivating domain complexes with the TATA box binding protein. Proc. Natl. Acad. Sci. USA 1994, 91, 2488–2492. [Google Scholar] [CrossRef] [PubMed]
- Boyer, T.G.; Martin, M.E.; Lees, E.; Ricciardi, R.P.; Berk, A.J. Mammalian Srb/Mediator complex is targeted by adenovirus E1A protein. Nature 1999, 399, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Vijayalingam, S.; Chinnadurai, G. Adenovirus L-E1A activates transcription through mediator complex-dependent recruitment of the super elongation complex. J. Virol. 2013, 87, 3425–3434. [Google Scholar] [CrossRef] [PubMed]
- Berk, A.J.; Boyer, T.G.; Kapanidis, A.N.; Ebright, R.H.; Kobayashi, N.N.; Horn, P.J.; Sullivan, S.M.; Koop, R.; Surby, M.A.; Triezenberg, S.J. Mechanisms of viral activators. Cold Spring Harb. Symp. Quant. Biol. 1998, 63, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Geisberg, J.V.; Chen, J.L.; Ricciardi, R.P. Subregions of the adenovirus E1A transactivation domain target multiple components of the TFIID complex. Mol. Cell Biol. 1995, 15, 6283–6290. [Google Scholar] [CrossRef] [PubMed]
- Mazzarelli, J.M.; Mengus, G.; Davidson, I.; Ricciardi, R.P. The transactivation domain of adenovirus E1A interacts with the C terminus of human TAF(II)135. J. Virol. 1997, 71, 7978–7983. [Google Scholar]
- Scholer, H.R.; Ciesiolka, T.; Gruss, P. A nexus between Oct-4 and E1A: Implications for gene regulation in embryonic stem cells. Cell 1991, 66, 291–304. [Google Scholar] [CrossRef]
- Liu, F.; Green, M.R. A specific member of the ATF transcription factor family can mediate transcription activation by the adenovirus E1a protein. Cell 1990, 61, 1217–1224. [Google Scholar] [CrossRef]
- Pelka, P.; Ablack, J.N.; Torchia, J.; Turnell, A.S.; Grand, R.J.; Mymryk, J.S. Transcriptional control by adenovirus E1A conserved region 3 via p300/CBP. Nucleic Acids Res. 2009, 37, 1095–1106. [Google Scholar] [CrossRef]
- Arany, Z.; Newsome, D.; Oldread, E.; Livingston, D.M.; Eckner, R. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature 1995, 374, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Eckner, R.; Ewen, M.E.; Newsome, D.; Gerdes, M.; DeCaprio, J.A.; Lawrence, J.B.; Livingston, D.M. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994, 8, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Marmorstein, R. Structure of the retinoblastoma protein bound to adenovirus E1A reveals the molecular basis for viral oncoprotein inactivation of a tumor suppressor. Genes Dev. 2007, 21, 2711–2716. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, J.C.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Structural basis for subversion of cellular control mechanisms by the adenoviral E1A oncoprotein. Proc. Natl. Acad. Sci. USA 2009, 106, 13260–13265. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Guida, P.; McCall, C.; Harlow, E. Adenovirus E1A makes two distinct contacts with the retinoblastoma protein. J. Virol. 1992, 66, 4606–4611. [Google Scholar] [PubMed]
- Berk, A.J. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 2005, 24, 7673–7685. [Google Scholar] [CrossRef] [PubMed]
- Schrier, P.I.; Bernards, R.; Vaessen, R.T.; Houweling, A.; van der Eb, A.J. Expression of class I major histocompatibility antigens switched off by highly oncogenic adenovirus 12 in transformed rat cells. Nature 1983, 305, 771–775. [Google Scholar] [CrossRef]
- Bernards, R.; Schrier, P.I.; Houweling, A.; Bos, J.L.; van der Eb, A.J.; Zijlstra, M.; Melief, C.J. Tumorigenicity of cells transformed by adenovirus type 12 by evasion of T-cell immunity. Nature 1983, 305, 776–779. [Google Scholar] [CrossRef]
- Gallimore, P.H.; Turnell, A.S. Adenovirus E1A: Remodelling the host cell, a life or death experience. Oncogene 2001, 20, 7824–7835. [Google Scholar] [CrossRef]
- Obenauer, J.C.; Denson, J.; Mehta, P.K.; Su, X.; Mukatira, S.; Finkelstein, D.B.; Xu, X.; Wang, J.; Ma, J.; Fan, Y.; et al. Large-scale sequence analysis of avian influenza isolates. Science 2006, 311, 1576–1580. [Google Scholar] [CrossRef]
- Martinez-Martin, N.; Ramani, S.R.; Hackney, J.A.; Tom, I.; Wranik, B.J.; Chan, M.; Wu, J.; Paluch, M.T.; Takeda, K.; Hass, P.E.; et al. The extracellular interactome of the human adenovirus family reveals diverse strategies for immunomodulation. Nat. Commun. 2016, 7, 11473. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Bielawski, J.P.; Yang, Z. A maximum likelihood method for detecting functional divergence at individual codon sites, with application to gene family evolution. J. Mol. Evol. 2004, 59, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Weadick, C.J.; Chang, B.S. An improved likelihood ratio test for detecting site-specific functional divergence among clades of protein-coding genes. Mol. Biol. Evol. 2012, 29, 1297–1300. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 2012, 80, 1715–1735. [Google Scholar] [CrossRef]
- Cilia, E.; Pancsa, R.; Tompa, P.; Lenaerts, T.; Vranken, W.F. From protein sequence to dynamics and disorder with DynaMine. Nat. Commun. 2013, 4, 2741. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Team, R.C. A Language and Environment for Statistical Computing. 2013. Available online: https://www.r-project.org/ (accessed on 3 February 2019).
- Le, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Robinson, C.M.; Rajaiya, J.; Walsh, M.P.; Seto, D.; Dyer, D.W.; Jones, M.S.; Chodosh, J. Computational analysis of human adenovirus type 22 provides evidence for recombination among species D human adenoviruses in the penton base gene. J. Virol. 2009, 83, 8980–8985. [Google Scholar] [CrossRef] [PubMed]
- Pelka, P.; Ablack, J.N.; Fonseca, G.J.; Yousef, A.F.; Mymryk, J.S. Intrinsic structural disorder in adenovirus E1A: A viral molecular hub linking multiple diverse processes. J. Virol. 2008, 82, 7252–7263. [Google Scholar] [CrossRef] [PubMed]
- Culp, J.S.; Webster, L.C.; Friedman, D.J.; Smith, C.L.; Huang, W.J.; Wu, F.Y.; Rosenberg, M.; Ricciardi, R.P. The 289-amino acid E1A protein of adenovirus binds zinc in a region that is important for trans-activation. Proc. Natl. Acad. Sci. USA 1988, 85, 6450–6454. [Google Scholar] [CrossRef] [PubMed]
- Kajan, G.L.; Kajon, A.E.; Pinto, A.C.; Bartha, D.; Arnberg, N. The complete genome sequence of human adenovirus 84, a highly recombinant new Human mastadenovirus D type with a unique fiber gene. Virus Res. 2017, 242, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.M.; Lee, J.S.; Dyer, D.W.; Seto, D.; Rajaiya, J.; Chodosh, J. Selection Pressure in the Human Adenovirus Fiber Knob Drives Cell Specificity in Epidemic Keratoconjunctivitis. J. Virol. 2016, 90, 9598–9607. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Seto, D.; Jones, M.S.; Dyer, D.W.; Chodosh, J. Molecular evolution of human species D adenoviruses. Infect. Genet. Evol. 2011, 11, 1208–1217. [Google Scholar] [CrossRef]
- Turnell, A.S.; Mymryk, J.S. Roles for the coactivators CBP and p300 and the APC/C E3 ubiquitin ligase in E1A-dependent cell transformation. Br. J. Cancer 2006, 95, 555–560. [Google Scholar] [CrossRef]
- Wevers, D.; Metzger, S.; Babweteera, F.; Bieberbach, M.; Boesch, C.; Cameron, K.; Couacy-Hymann, E.; Cranfield, M.; Gray, M.; Harris, L.A.; et al. Novel adenoviruses in wild primates: A high level of genetic diversity and evidence of zoonotic transmissions. J. Virol. 2011, 85, 10774–10784. [Google Scholar] [CrossRef]
- Chen, E.C.; Yagi, S.; Kelly, K.R.; Mendoza, S.P.; Tarara, R.P.; Canfield, D.R.; Maninger, N.; Rosenthal, A.; Spinner, A.; Bales, K.L.; et al. Cross-species transmission of a novel adenovirus associated with a fulminant pneumonia outbreak in a new world monkey colony. PLoS Pathog. 2011, 7, e1002155. [Google Scholar] [CrossRef]
- Dehghan, S.; Seto, J.; Jones, M.S.; Dyer, D.W.; Chodosh, J.; Seto, D. Simian adenovirus type 35 has a recombinant genome comprising human and simian adenovirus sequences, which predicts its potential emergence as a human respiratory pathogen. Virology 2013, 447, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Purkayastha, A.; Ditty, S.E.; Su, J.; McGraw, J.; Hadfield, T.L.; Tibbetts, C.; Seto, D. Genomic and bioinformatics analysis of HAdV-4, a human adenovirus causing acute respiratory disease: Implications for gene therapy and vaccine vector development. J. Virol. 2005, 79, 2559–2572. [Google Scholar] [CrossRef] [PubMed]
- Gersbach, C.A.; Gaj, T.; Barbas, C.F., 3rd. Synthetic zinc finger proteins: The advent of targeted gene regulation and genome modification technologies. Acc. Chem. Res. 2014, 47, 2309–2318. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Full E1A-13S Nucleotide Sequence | |||||||
|---|---|---|---|---|---|---|---|
| Model | Np | ln L | κ | Null | LR | df | p |
| CmC_2partition_Dcombined | 123 | −7622.25 | 2.21 | ||||
| CmC_Alternate_3partition_groups1_2 | 124 | −7589.64 | 1.98 | CmC_Dcombined_2partition | 65.22 | 1 | 6.71 × 10−16 |
| 62AA CR3 sequence removed from full E1A-13S | |||||||
| Model | Np | ln L | κ | Null | LR | df | p |
| CmC_2partition_Dcombined | 123 | −5826.58 | 2.06 | ||||
| CmC_Alternate_3partition_groups1_2 | 124 | −5825.29 | 2.05 | CmC_Dcombined_2partition | 2.58 | 1 | 0.108 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, G.; Ismail, A.M.; Lee, J.Y.; Ramke, M.; Lee, J.S.; Dyer, D.W.; Seto, D.; Rajaiya, J.; Chodosh, J. Divergent Evolution of E1A CR3 in Human Adenovirus Species D. Viruses 2019, 11, 143. https://doi.org/10.3390/v11020143
Singh G, Ismail AM, Lee JY, Ramke M, Lee JS, Dyer DW, Seto D, Rajaiya J, Chodosh J. Divergent Evolution of E1A CR3 in Human Adenovirus Species D. Viruses. 2019; 11(2):143. https://doi.org/10.3390/v11020143
Chicago/Turabian StyleSingh, Gurdeep, Ashrafali M. Ismail, Jeong Yoon Lee, Mirja Ramke, Ji Sun Lee, David W. Dyer, Donald Seto, Jaya Rajaiya, and James Chodosh. 2019. "Divergent Evolution of E1A CR3 in Human Adenovirus Species D" Viruses 11, no. 2: 143. https://doi.org/10.3390/v11020143
APA StyleSingh, G., Ismail, A. M., Lee, J. Y., Ramke, M., Lee, J. S., Dyer, D. W., Seto, D., Rajaiya, J., & Chodosh, J. (2019). Divergent Evolution of E1A CR3 in Human Adenovirus Species D. Viruses, 11(2), 143. https://doi.org/10.3390/v11020143