Angiomotin-Like 1 Links Paramyxovirus M Proteins to NEDD4 Family Ubiquitin Ligases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
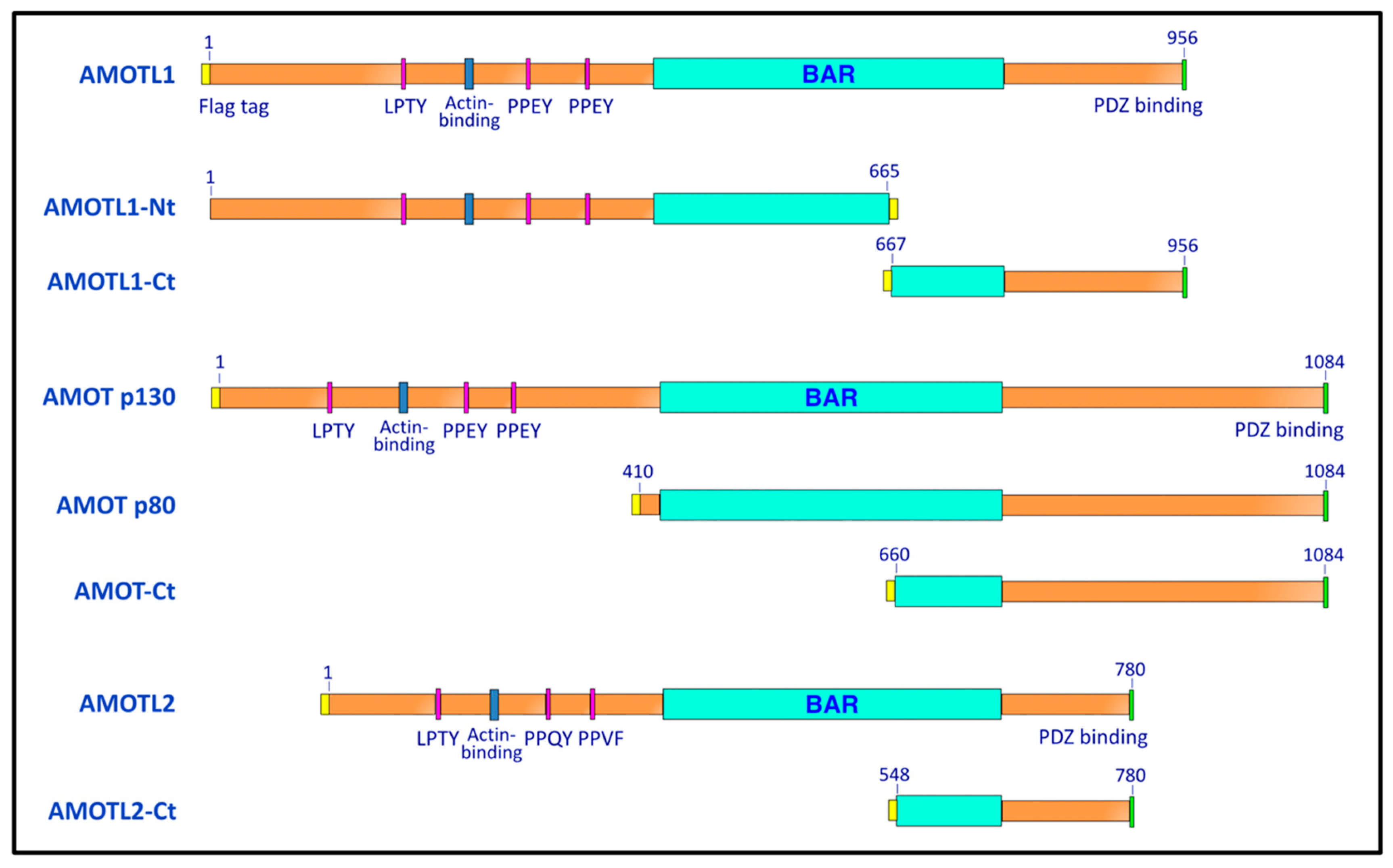
2.1. Plasmids
2.2. Antibodies
2.3. Co-Immunoprecipitation
2.4. Measurements of VLP Production
3. Results
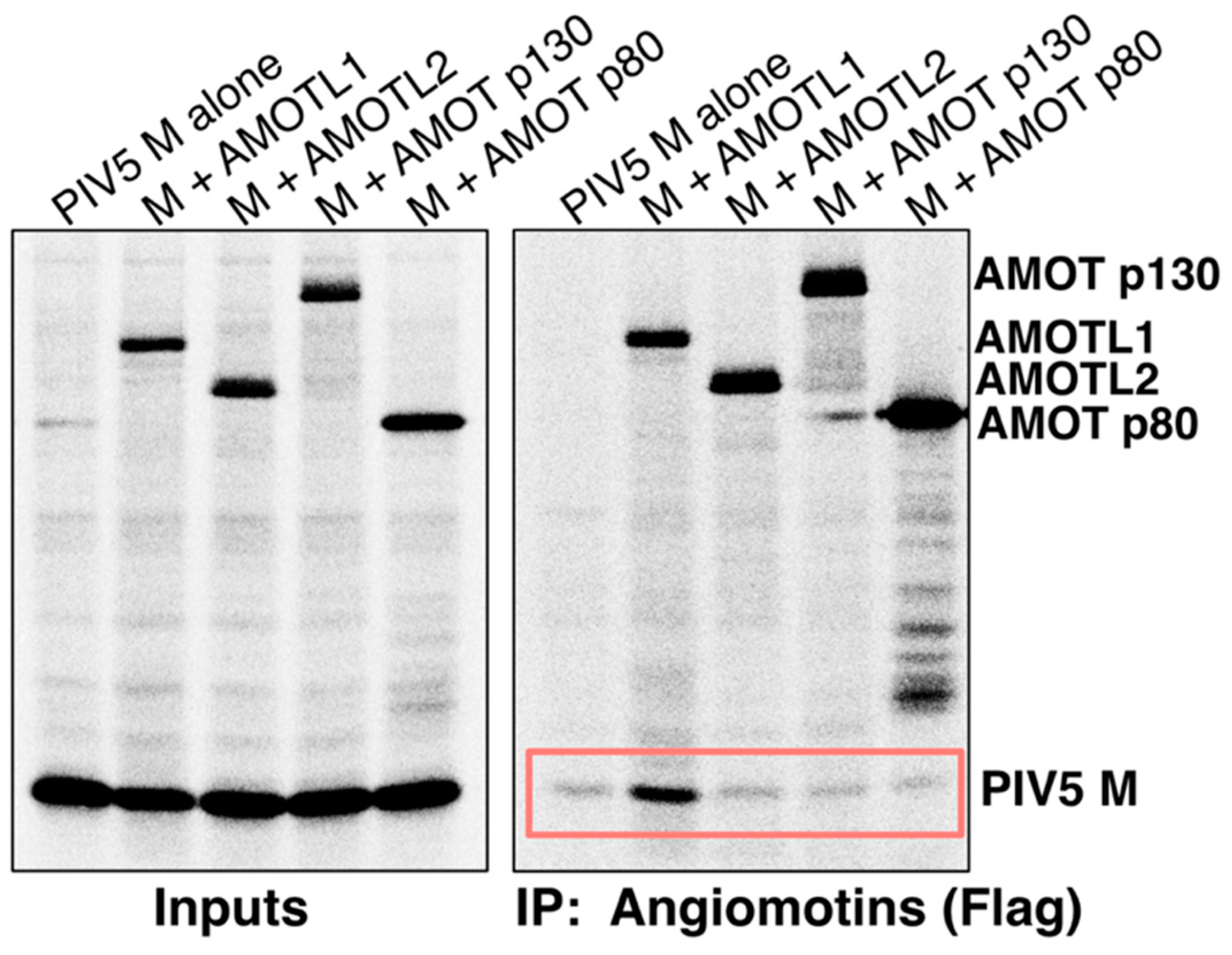
3.1. Paramyxovirus M Proteins Bind Selectively to AMOTL1
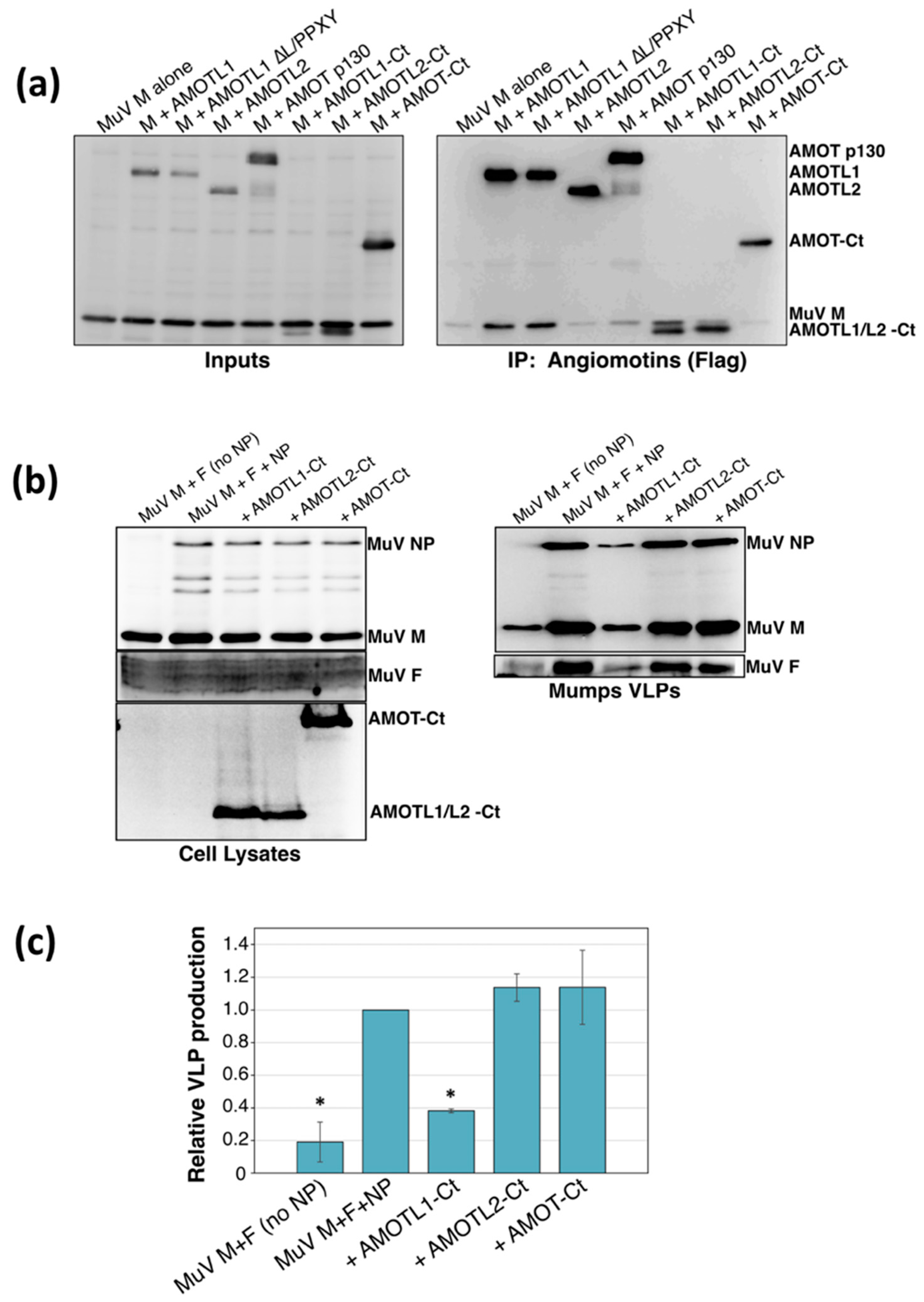
3.2. AMOTL1 Binds to Multiple NEDD4 Family Proteins via L/PPXY Motifs
3.3. AMOTL1 Links NEDD4 Family Proteins to PIV5 M
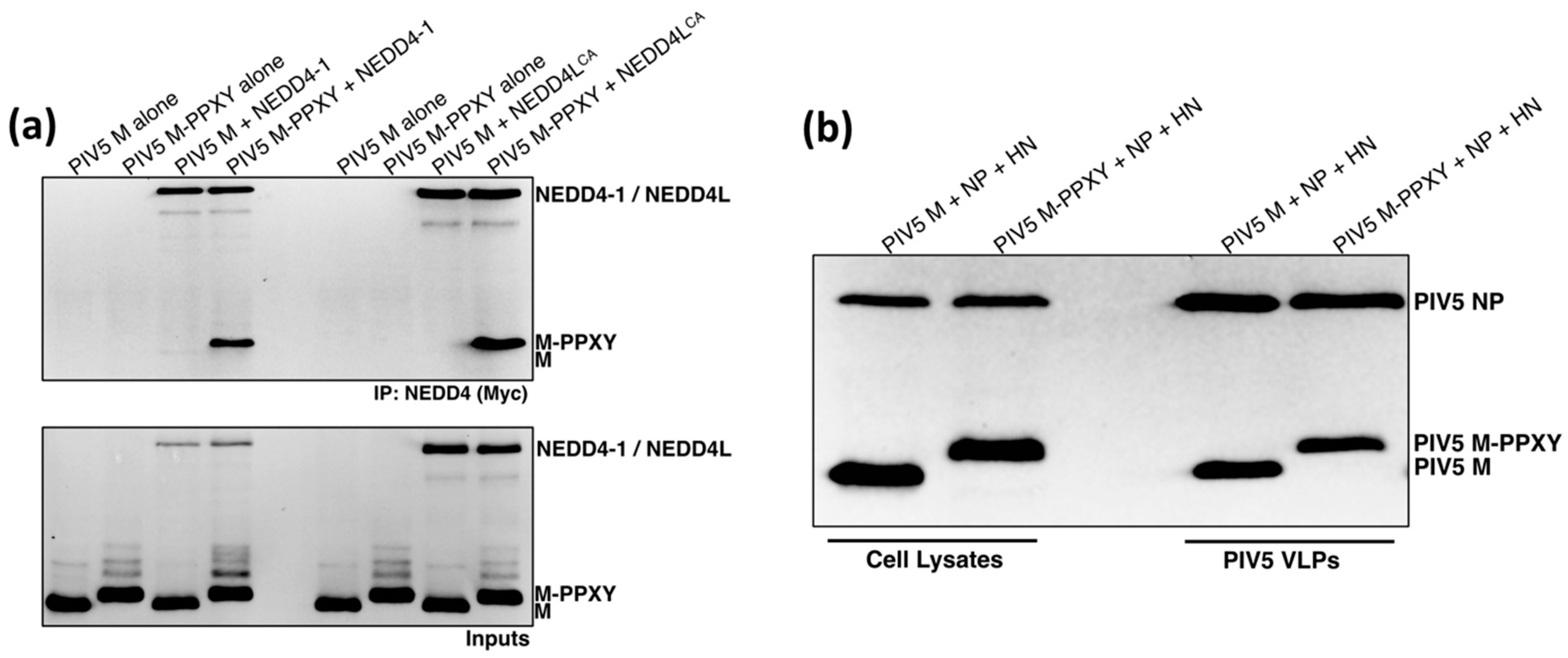
3.4. PIV5 M-PPXY Binds to NEDD4L without the Need for AMOTL1
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lamb, R.A.; Parks, G.D. Paramyxoviridae: The viruses and their replication. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P., Eds.; Williams and Wilkins: Philadelphia, PA, USA, 2013; pp. 957–995. [Google Scholar]
- Cox, R.M.; Plemper, R.K. Structure and organization of paramyxovirus particles. Curr. Opin. Virol. 2017, 24, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.S.; Sakaguchi, T.; Schmitt, A.P. Paramyxovirus assembly and budding: Building particles that transmit infections. Int. J. Biochem. Cell Biol. 2010, 42, 1416–1429. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Weissenhorn, W.; Poudevigne, E.; Effantin, G.; Bassereau, P. How to get out: ssRNA enveloped viruses and membrane fission. Curr. Opin. Virol. 2013, 3, 159–167. [Google Scholar] [CrossRef]
- Bieniasz, P.D. Late budding domains and host proteins in enveloped virus release. Virology 2006, 344, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Macias, M.J.; Wiesner, S.; Sudol, M. WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 2002, 513, 30–37. [Google Scholar] [CrossRef]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef]
- Craven, R.C.; Harty, R.N.; Paragas, J.; Palese, P.; Wills, J.W. Late domain function identified in the vesicular stomatitis virus M protein by use of rhabdovirus-retrovirus chimeras. J. Virol. 1999, 73, 3359–3365. [Google Scholar]
- Harty, R.N.; Paragas, J.; Sudol, M.; Palese, P. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: Implications for viral budding. J. Virol. 1999, 73, 2921–2929. [Google Scholar]
- Jayakar, H.R.; Murti, K.G.; Whitt, M.A. Mutations in the PPPY motif of vesicular stomatitis virus matrix protein reduce virus budding by inhibiting a late step in virion release. J. Virol. 2000, 74, 9818–9827. [Google Scholar] [CrossRef]
- Harty, R.N.; Brown, M.E.; Wang, G.; Huibregtse, J.M.; Hayes, F.P. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: Implications for filovirus budding. Proc. Natl. Acad. Sci. USA 2000, 97, 13871–13876. [Google Scholar] [CrossRef]
- Licata, J.M.; Simpson-Holley, M.; Wright, N.T.; Han, Z.; Paragas, J.; Harty, R.N. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: Involvement of host proteins TSG101 and VPS-4. J. Virol. 2003, 77, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef]
- Perez, M.; Craven, R.C.; de la Torre, J.C. The small RING finger protein Z drives arenavirus budding: Implications for antiviral strategies. Proc. Natl. Acad. Sci. USA 2003, 100, 12978–12983. [Google Scholar] [CrossRef] [PubMed]
- Wirblich, C.; Tan, G.S.; Papaneri, A.; Godlewski, P.J.; Orenstein, J.M.; Harty, R.N.; Schnell, M.J. PPEY motif within the rabies virus (RV) matrix protein is essential for efficient virion release and RV pathogenicity. J. Virol. 2008, 82, 9730–9738. [Google Scholar] [CrossRef]
- Li, M.; Schmitt, P.T.; Li, Z.; McCrory, T.S.; He, B.; Schmitt, A.P. Mumps virus matrix, fusion, and nucleocapsid proteins cooperate for efficient production of virus-like particles. J. Virol. 2009, 83, 7261–7272. [Google Scholar] [CrossRef]
- Schmitt, A.P.; Leser, G.P.; Morita, E.; Sundquist, W.I.; Lamb, R.A. Evidence for a new viral late-domain core sequence, FPIV, necessary for budding of a paramyxovirus. J. Virol. 2005, 79, 2988–2997. [Google Scholar] [CrossRef]
- Duan, Z.; Hu, Z.; Zhu, J.; Xu, H.; Chen, J.; Liu, H.; Hu, S.; Liu, X. Mutations in the FPIV motif of Newcastle disease virus matrix protein attenuate virus replication and reduce virus budding. Arch. Virol. 2014, 159, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Irie, T.; Nagata, N.; Yoshida, T.; Sakaguchi, T. Recruitment of Alix/AIP1 to the plasma membrane by Sendai virus C protein facilitates budding of virus-like particles. Virology 2008, 371, 108–120. [Google Scholar] [CrossRef]
- Sakaguchi, T.; Kato, A.; Sugahara, F.; Shimazu, Y.; Inoue, M.; Kiyotani, K.; Nagai, Y.; Yoshida, T. AIP1/Alix is a binding partner of Sendai virus C protein and facilitates virus budding. J. Virol. 2005, 79, 8933–8941. [Google Scholar] [CrossRef]
- Boonyaratanakornkit, J.; Schomacker, H.; Collins, P.; Schmidt, A. Alix serves as an adaptor that allows human parainfluenza virus type 1 to interact with the host cell ESCRT system. PLoS ONE 2013, 8, e59462. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Yun, T.; Vigant, F.; Pernet, O.; Won, S.T.; Dawes, B.E.; Bartkowski, W.; Freiberg, A.N.; Lee, B. Nipah Virus C Protein Recruits Tsg101 to Promote the Efficient Release of Virus in an ESCRT-Dependent Pathway. PLoS Pathog. 2016, 12, e1005659. [Google Scholar] [CrossRef]
- Pei, Z.; Bai, Y.; Schmitt, A.P. PIV5 M protein interaction with host protein angiomotin-like 1. Virology 2010, 397, 155–166. [Google Scholar] [CrossRef]
- Moleirinho, S.; Guerrant, W.; Kissil, J.L. The Angiomotins--from discovery to function. FEBS Lett. 2014, 588, 2693–2703. [Google Scholar] [CrossRef] [PubMed]
- Bratt, A.; Birot, O.; Sinha, I.; Veitonmäki, N.; Aase, K.; Ernkvist, M.; Holmgren, L. Angiomotin regulates endothelial cell-cell junctions and cell motility. J. Biol. Chem. 2005, 280, 34859–34869. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Vertuani, S.; Nyström, S.; Audebert, S.; Meijer, I.; Tegnebratt, T.; Borg, J.P.; Uhlén, P.; Majumdar, A.; Holmgren, L. Angiomotin-like protein 1 controls endothelial polarity and junction stability during sprouting angiogenesis. Circ. Res. 2009, 105, 260–270. [Google Scholar] [CrossRef]
- Nishimura, M.; Kakizaki, M.; Ono, Y.; Morimoto, K.; Takeuchi, M.; Inoue, Y.; Imai, T.; Takai, Y. JEAP, a novel component of tight junctions in exocrine cells. J. Biol. Chem. 2002, 277, 5583–5587. [Google Scholar] [CrossRef]
- Sugihara-Mizuno, Y.; Adachi, M.; Kobayashi, Y.; Hamazaki, Y.; Nishimura, M.; Imai, T.; Furuse, M.; Tsukita, S. Molecular characterization of angiomotin/JEAP family proteins: Interaction with MUPP1/Patj and their endogenous properties. Genes Cells 2007, 12, 473–486. [Google Scholar] [CrossRef]
- Wells, C.D.; Fawcett, J.P.; Traweger, A.; Yamanaka, Y.; Goudreault, M.; Elder, K.; Kulkarni, S.; Gish, G.; Virag, C.; Lim, C.; et al. A Rich1/Amot complex regulates the Cdc42 GTPase and apical-polarity proteins in epithelial cells. Cell 2006, 125, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Shen, Z.; Stemmer-Rachamimov, A.; Dawany, N.; Troutman, S.; Showe, L.C.; Liu, Q.; Shimono, A.; Sudol, M.; Holmgren, L. The p130 isoform of angiomotin is required for Yap-mediated hepatic epithelial cell proliferation and tumorigenesis. Sci. Signal. 2013, 6, ra77. [Google Scholar] [CrossRef] [PubMed]
- Mercenne, G.; Alam, S.L.; Arii, J.; Lalonde, M.S.; Sundquist, W.I. Angiomotin functions in HIV-1 assembly and budding. eLife 2015, 4, e03778. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.-Y.; Morita, E.; von Schwedler, U.; Müller, B.; Kräusslich, H.-G.; Sundquist, W.I. NEDD4L overexpression rescues the release and infectivity of human immunodeficiency virus type 1 constructs lacking PTAP and YPXL late domains. J. Virol. 2008, 82, 4884–4897. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Popov, S.; Popova, E.; Göttlinger, H.G. Efficient and specific rescue of human immunodeficiency virus type 1 budding defects by a Nedd4-like ubiquitin ligase. J. Virol. 2008, 82, 4898–4907. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.P.; Leser, G.P.; Waning, D.L.; Lamb, R.A. Requirements for budding of paramyxovirus simian virus 5 virus-like particles. J. Virol. 2002, 76, 3952–3964. [Google Scholar] [CrossRef] [PubMed]
- Niwa, H.; Yamamura, K.; Miyazaki, J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 1991, 108, 193–199. [Google Scholar] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Randall, R.E.; Young, D.F.; Goswami, K.K.; Russell, W.C. Isolation and characterization of monoclonal antibodies to simian virus 5 and their use in revealing antigenic differences between human, canine and simian isolates. J. Gen. Virol. 1987, 68, 2769–2780. [Google Scholar] [CrossRef]
- Schmitt, P.T.; Ray, G.; Schmitt, A.P. The C-terminal end of parainfluenza virus 5 NP protein is important for virus-like particle production and M-NP protein interaction. J. Virol. 2010, 84, 12810–12823. [Google Scholar] [CrossRef] [PubMed]
- Skouloudaki, K.; Walz, G. YAP1 recruits c-Abl to protect angiomotin-like 1 from Nedd4-mediated degradation. PLoS ONE 2012, 7, e35735. [Google Scholar] [CrossRef]
- Choi, K.S.; Choi, H.J.; Lee, J.K.; Im, S.; Zhang, H.; Jeong, Y.; Park, J.A.; Lee, I.K.; Kim, Y.M.; Kwon, Y.G. The endothelial E3 ligase HECW2 promotes endothelial cell junctions by increasing AMOTL1 protein stability via K63-linked ubiquitination. Cell Signal. 2016, 28, 1642–1651. [Google Scholar] [CrossRef]
- Harty, R.N. No exit: Targeting the budding process to inhibit filovirus replication. Antivir. Res. 2009, 81, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.; Ebihara, H.; Groseth, A. Arenavirus budding: A common pathway with mechanistic differences. Viruses 2013, 5, 528–549. [Google Scholar] [CrossRef] [PubMed]
- Shields, S.B.; Piper, R.C. How ubiquitin functions with ESCRTs. Traffic 2011, 12, 1306–1317. [Google Scholar] [CrossRef] [PubMed]
- Garrus, J.E.; Von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Côté, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Harrison, M.S.; Schmitt, P.T.; Pei, Z.; Schmitt, A.P. Role of ubiquitin in parainfluenza virus 5 particle formation. J. Virol. 2012, 86, 3474–3485. [Google Scholar] [CrossRef] [PubMed]
- Pentecost, M.; Vashisht, A.A.; Lester, T.; Voros, T.; Beaty, S.M.; Park, A.; Wang, Y.E.; Yun, T.E.; Freiberg, A.N.; Wohlschlegel, J.A.; et al. Evidence for ubiquitin-regulated nuclear and subnuclear trafficking among Paramyxovirinae matrix proteins. PLoS Pathog. 2015, 11, e1004739. [Google Scholar] [CrossRef]
- Wang, Y.E.; Park, A.; Lake, M.; Pentecost, M.; Torres, B.; Yun, T.E.; Wolf, M.C.; Holbrook, M.R.; Freiberg, A.N.; Lee, B.; et al. Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog. 2010, 6, e1001186. [Google Scholar] [CrossRef] [PubMed]
- Mana-Capelli, S.; Paramasivam, M.; Dutta, S.; McCollum, D. Angiomotins link F-actin architecture to Hippo pathway signaling. Mol. Biol. Cell 2014, 25, 1676–1685. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ray, G.; Schmitt, P.T.; Schmitt, A.P. Angiomotin-Like 1 Links Paramyxovirus M Proteins to NEDD4 Family Ubiquitin Ligases. Viruses 2019, 11, 128. https://doi.org/10.3390/v11020128
Ray G, Schmitt PT, Schmitt AP. Angiomotin-Like 1 Links Paramyxovirus M Proteins to NEDD4 Family Ubiquitin Ligases. Viruses. 2019; 11(2):128. https://doi.org/10.3390/v11020128
Chicago/Turabian StyleRay, Greeshma, Phuong Tieu Schmitt, and Anthony P. Schmitt. 2019. "Angiomotin-Like 1 Links Paramyxovirus M Proteins to NEDD4 Family Ubiquitin Ligases" Viruses 11, no. 2: 128. https://doi.org/10.3390/v11020128
APA StyleRay, G., Schmitt, P. T., & Schmitt, A. P. (2019). Angiomotin-Like 1 Links Paramyxovirus M Proteins to NEDD4 Family Ubiquitin Ligases. Viruses, 11(2), 128. https://doi.org/10.3390/v11020128