Genomic Characterization of Orf Virus Strain D1701-V (Parapoxvirus) and Development of Novel Sites for Multiple Transgene Expression

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus
2.2. DNA Preparation, Restriction Digests, Southern Blotting
2.3. DNA Sequencing
2.4. Novel Synthetic ORFV Early Promoter eP1 and eP2
2.5. Construction of Transfer Plasmids
2.6. Nucleofection
2.7. Western Blotting
2.8. Flow Cytometry
2.9. Polymerase Chain Reaction (PCR)
2.10. Selection of ORFV Recombinants
3. Results
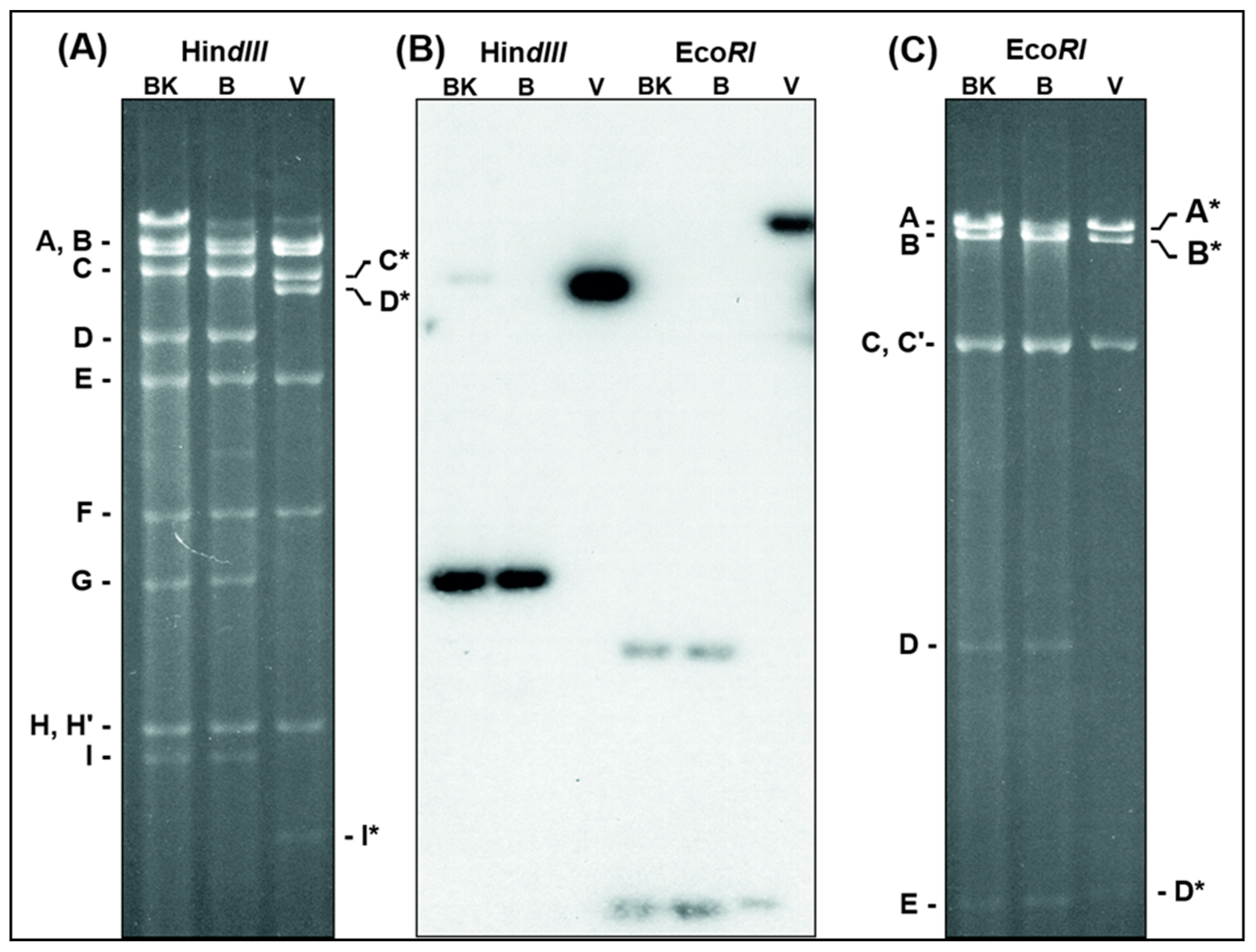
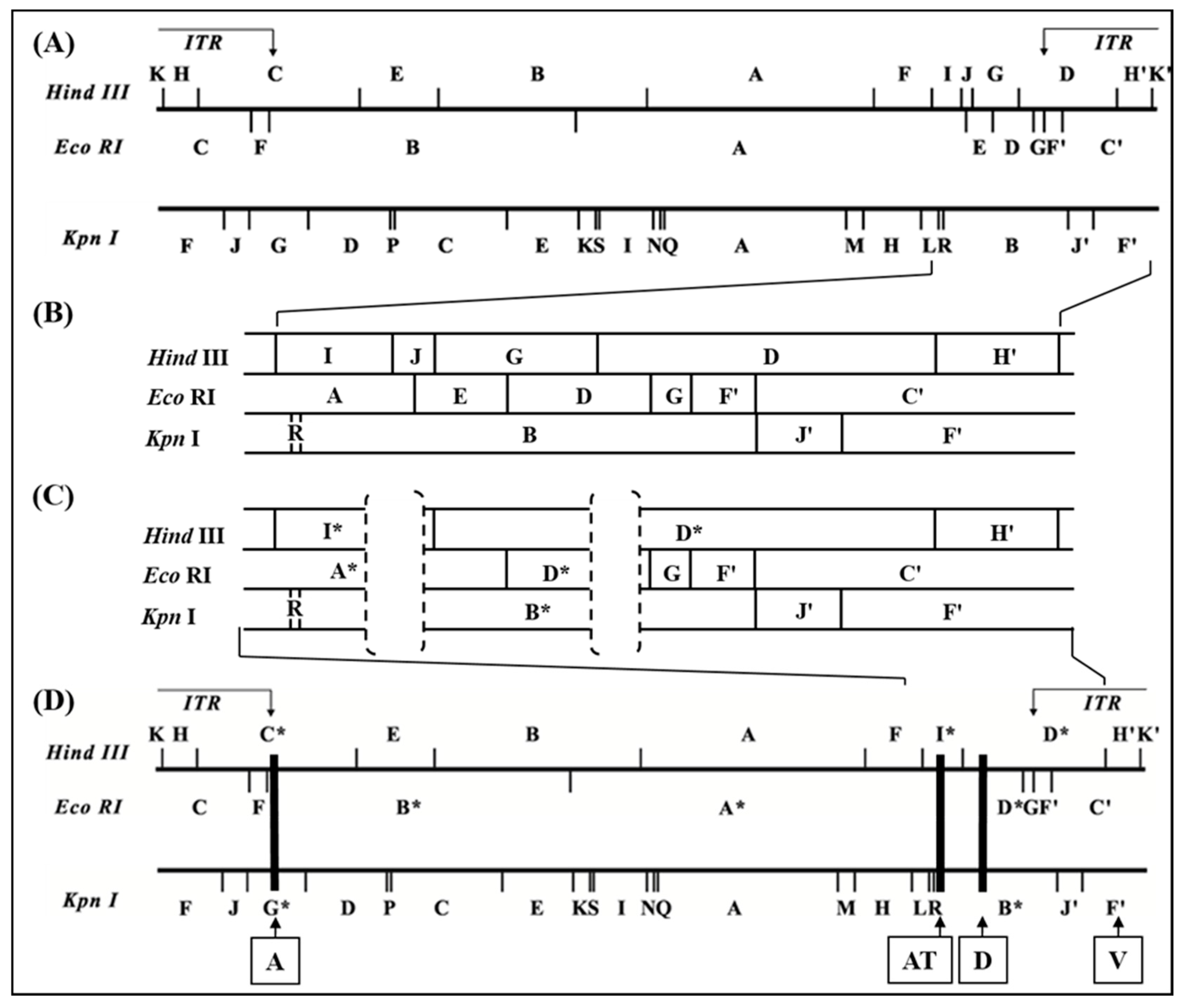
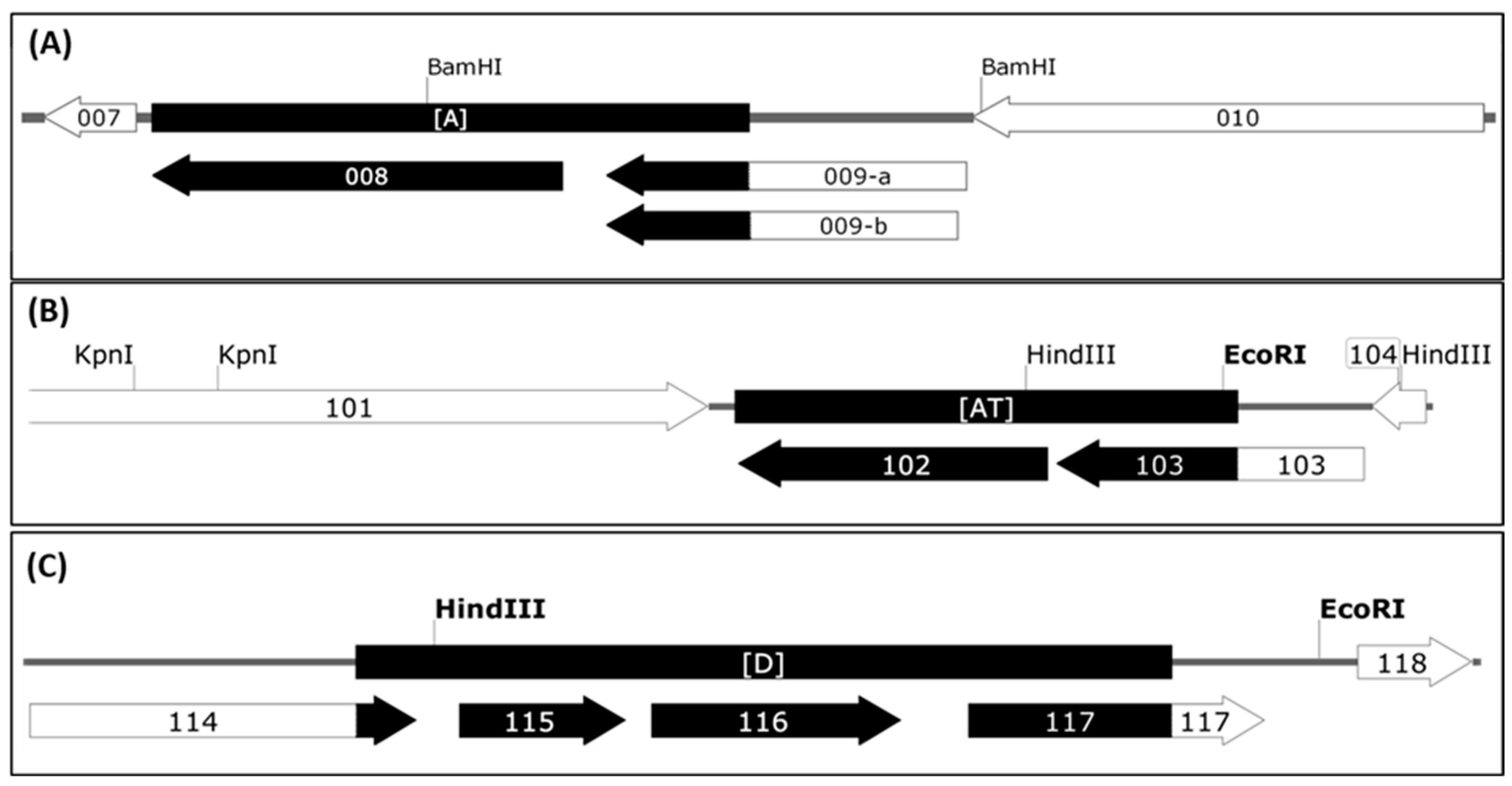
3.1. Detection and Mapping of Three Major Deletions in the D1701-V Genome.
3.2. ORFV Genes Affected by the 3 Deletions
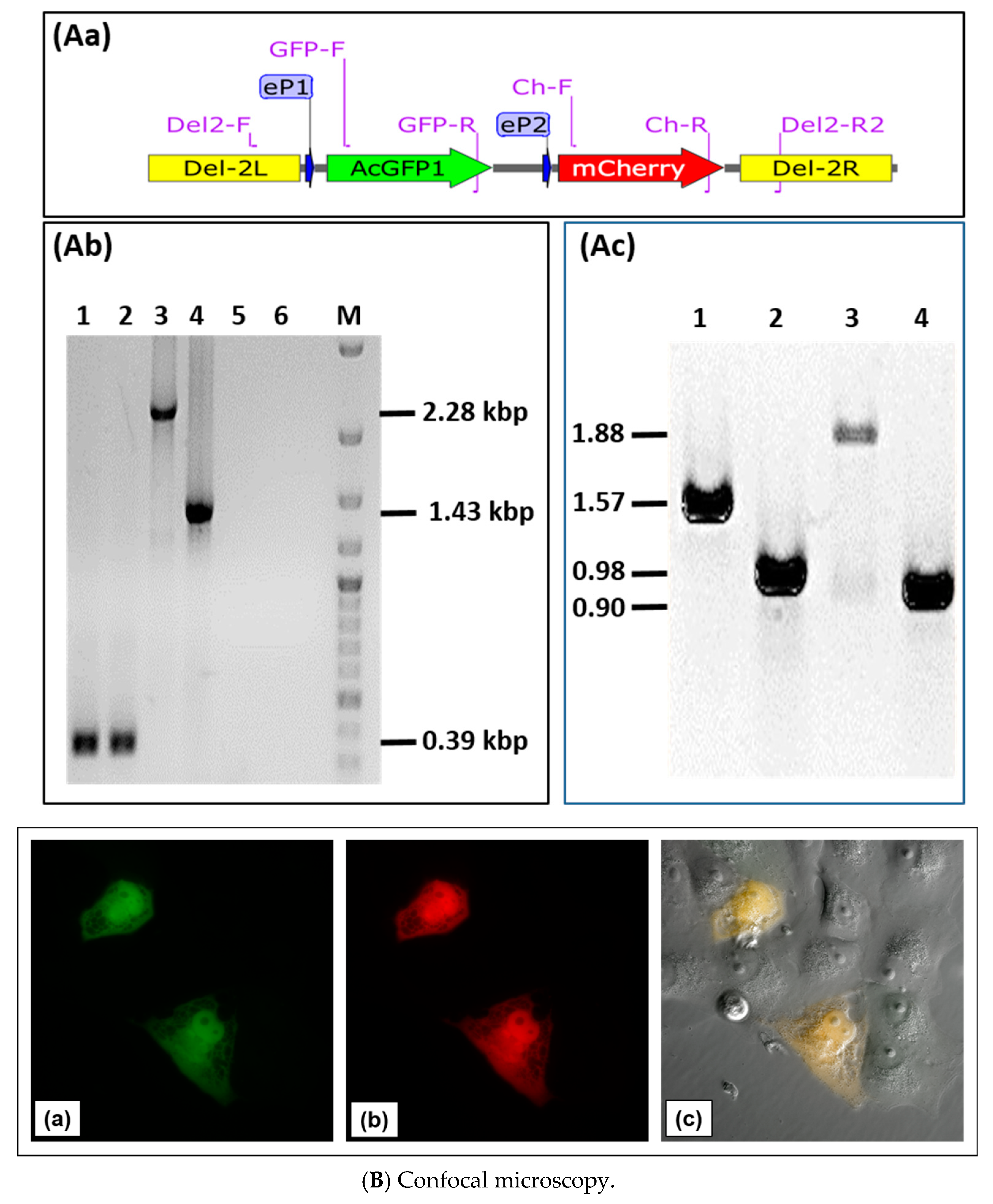
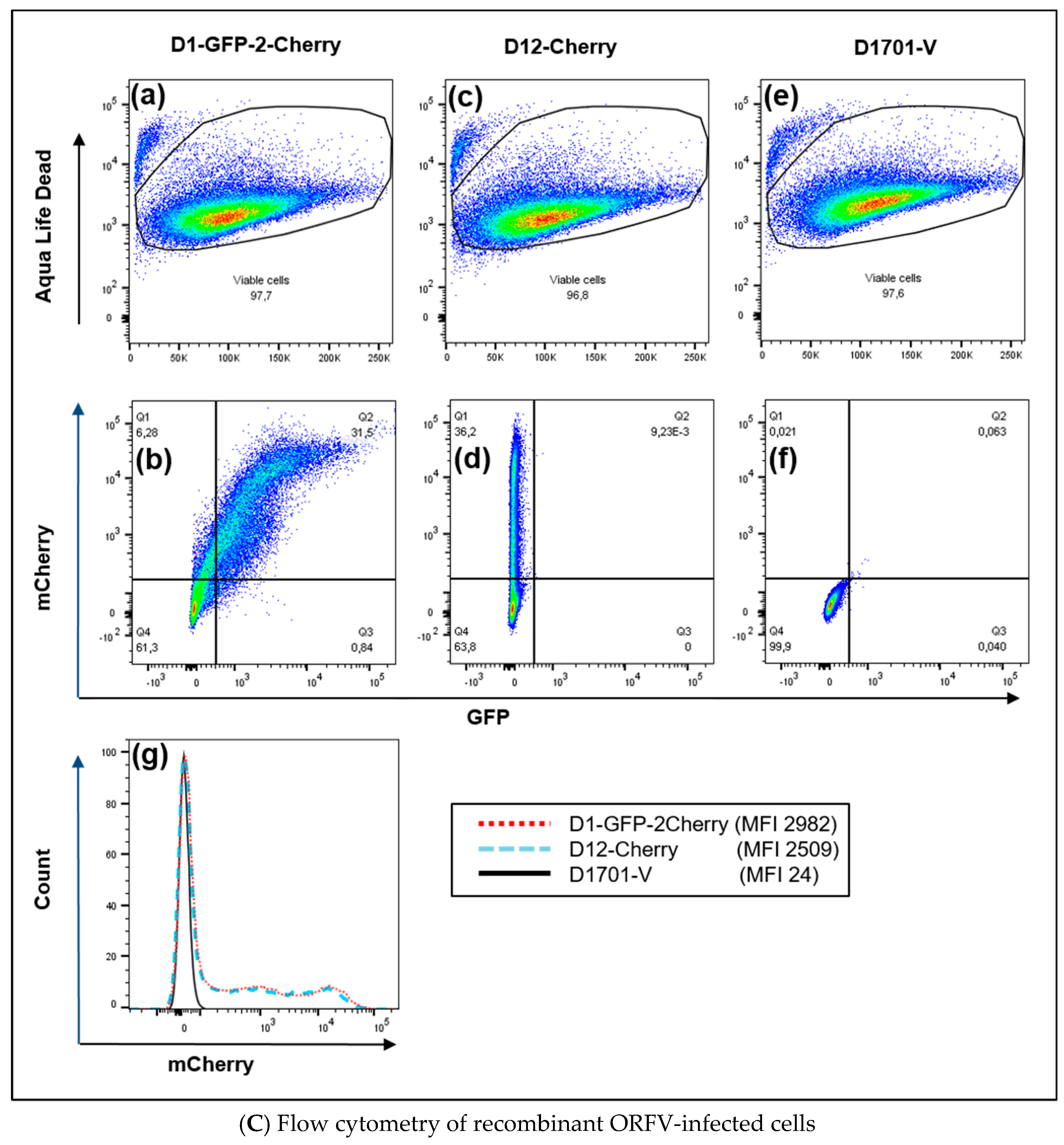
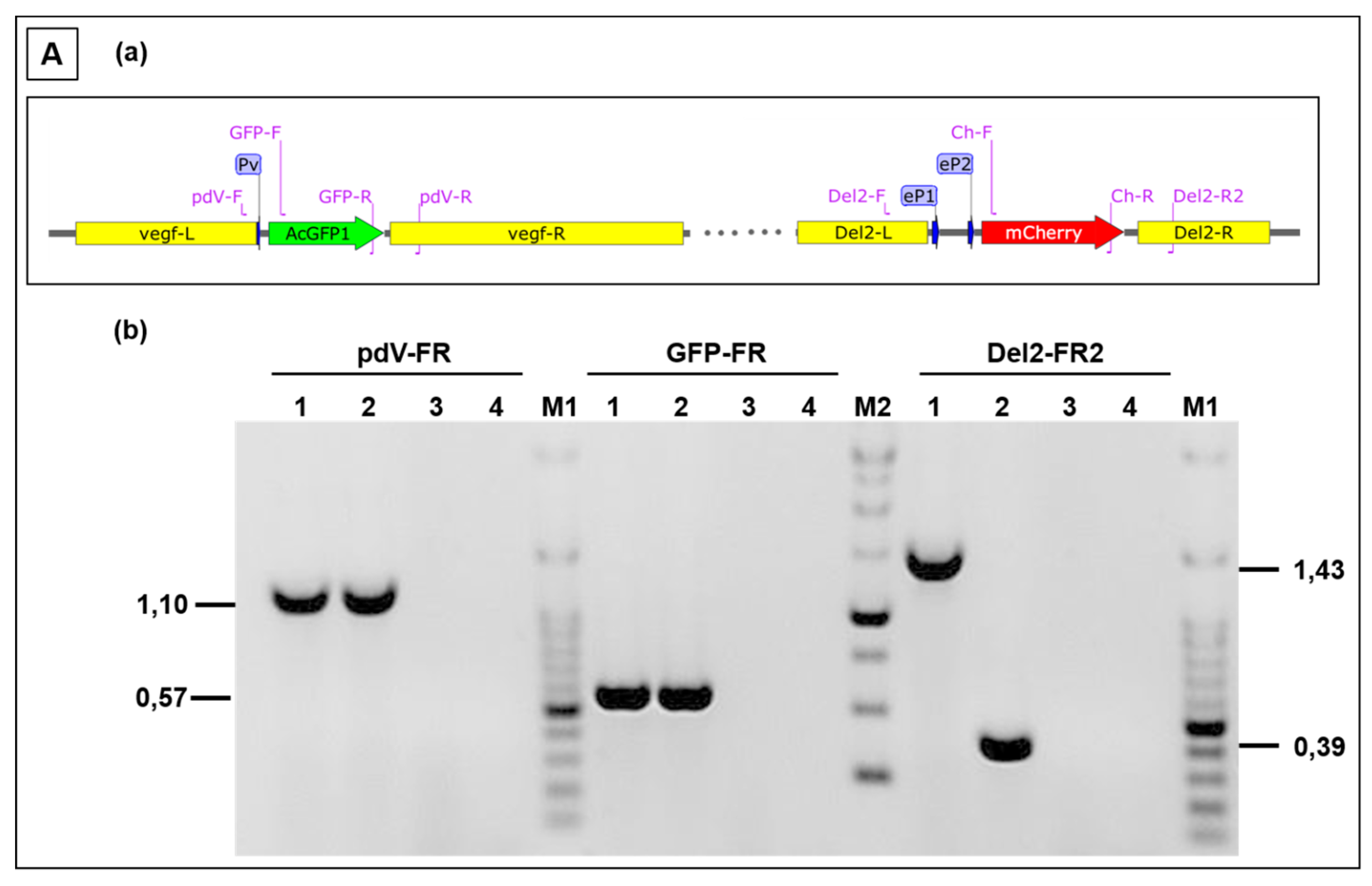
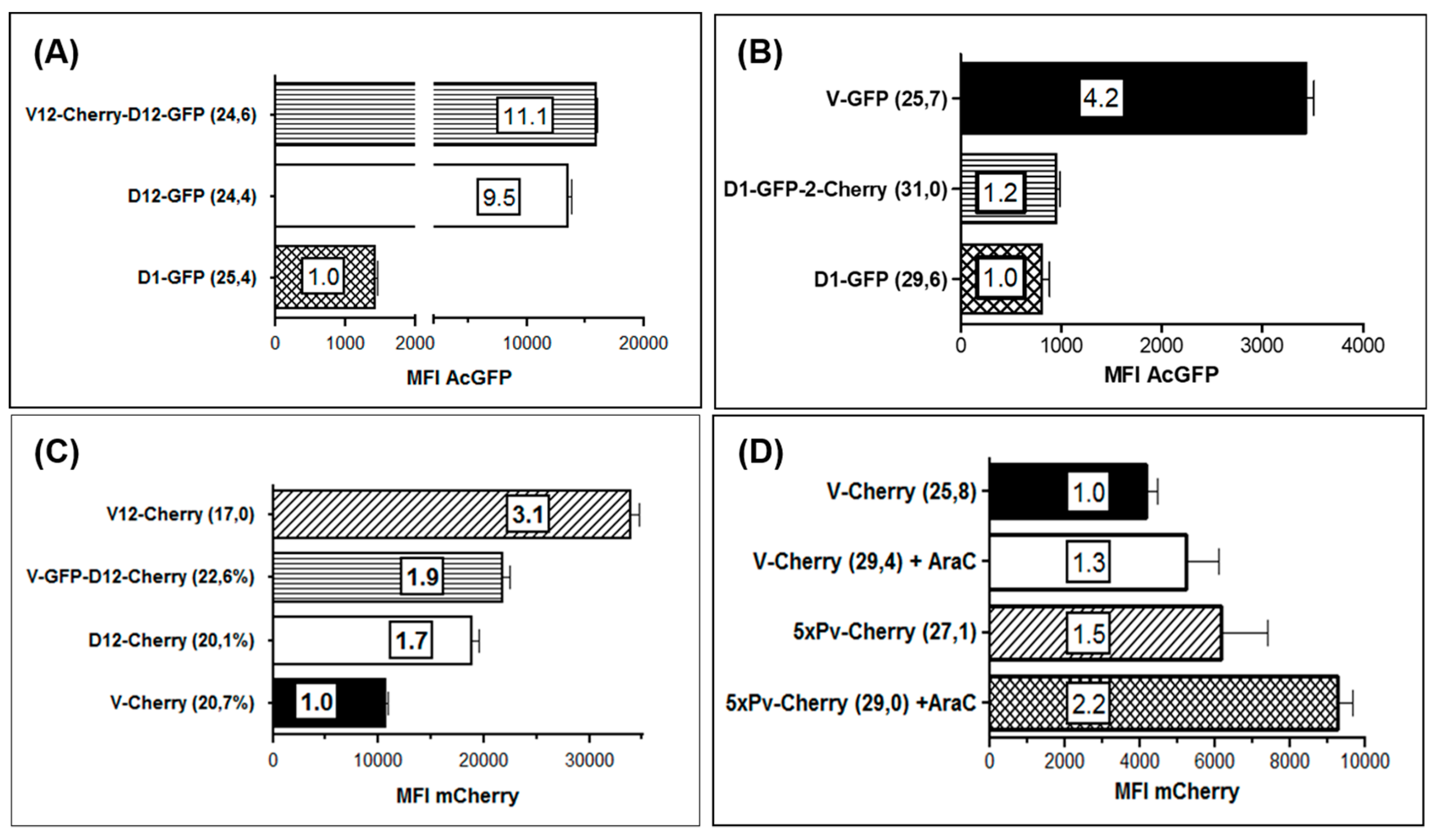
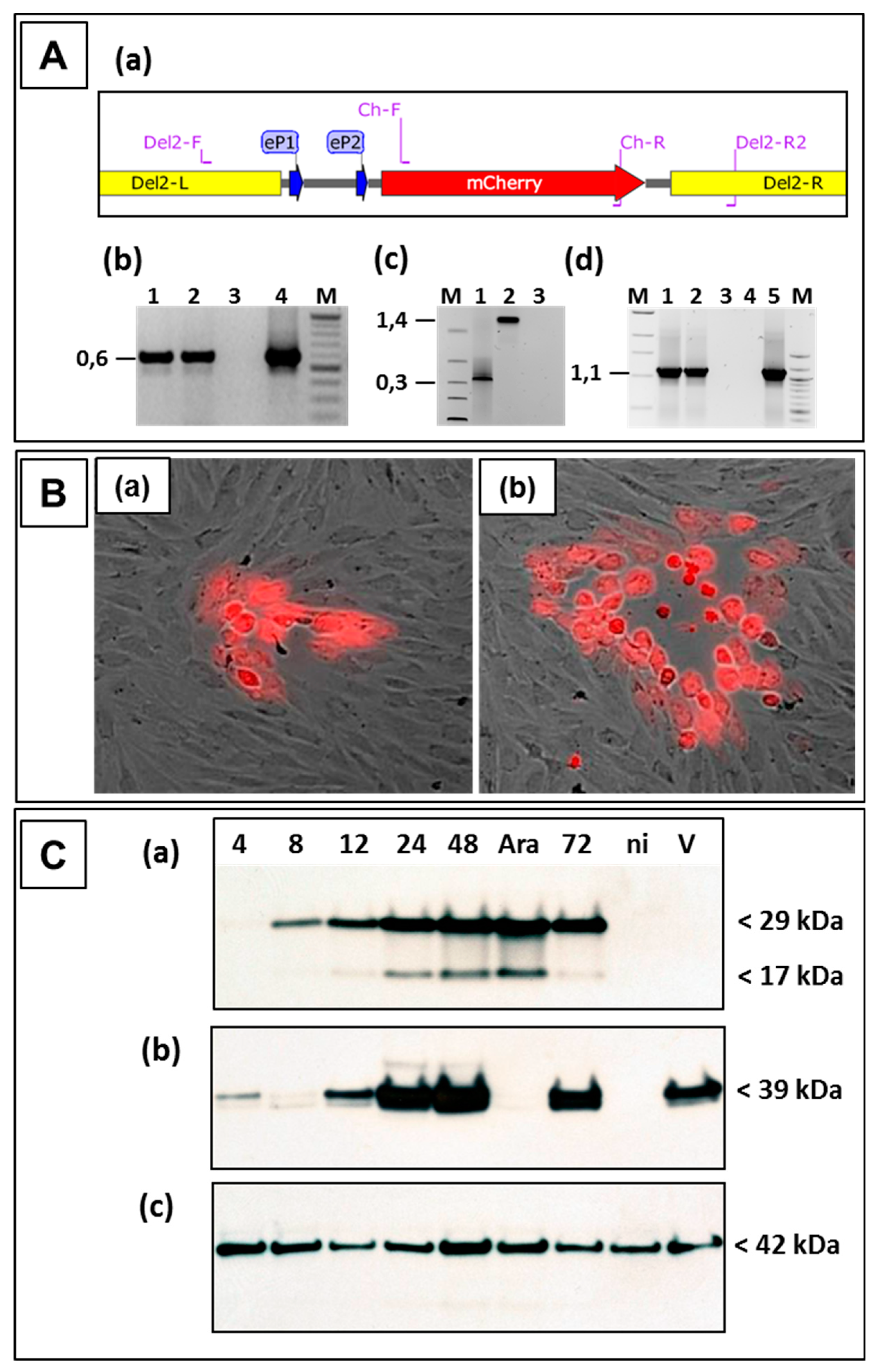
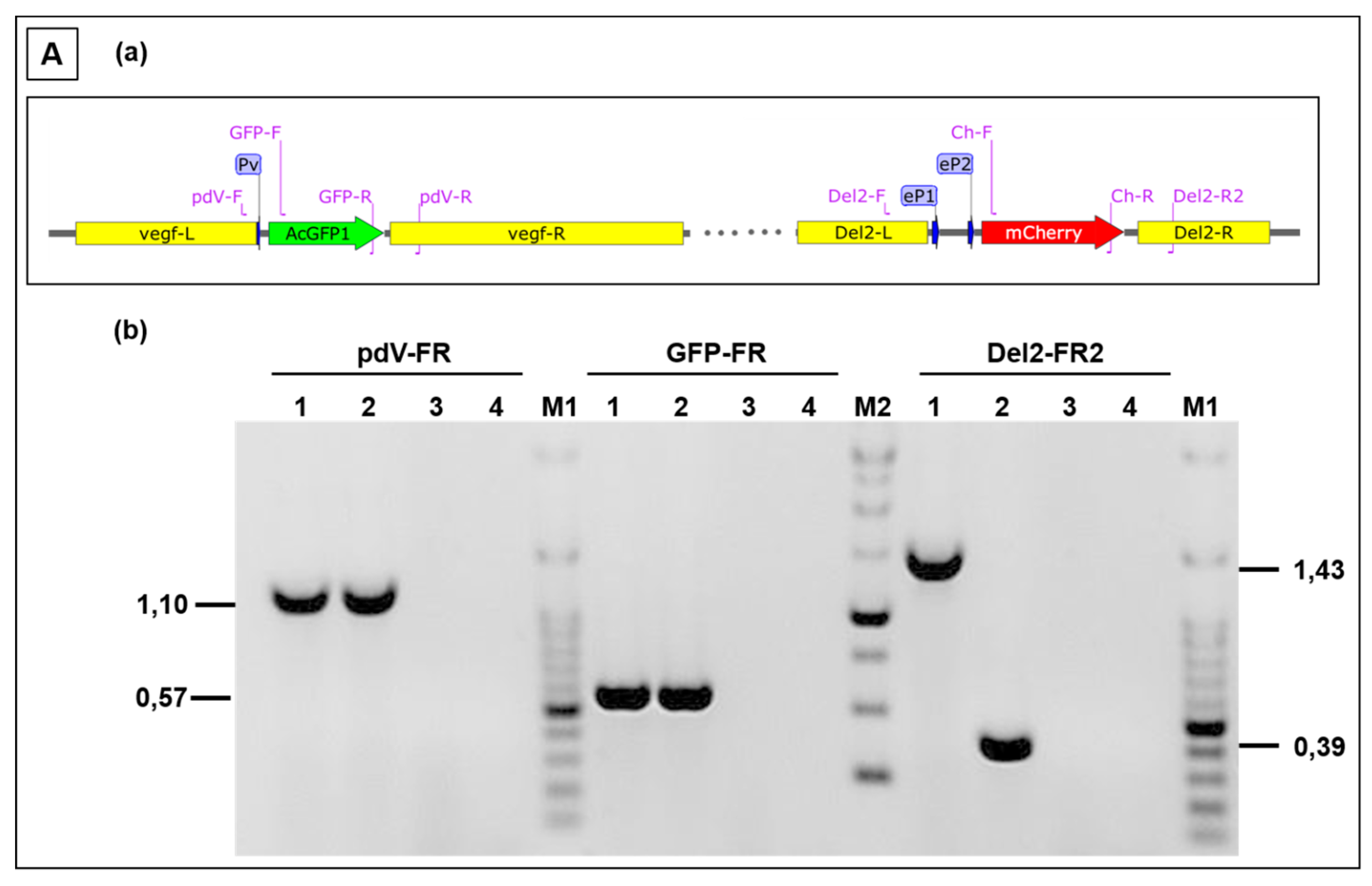
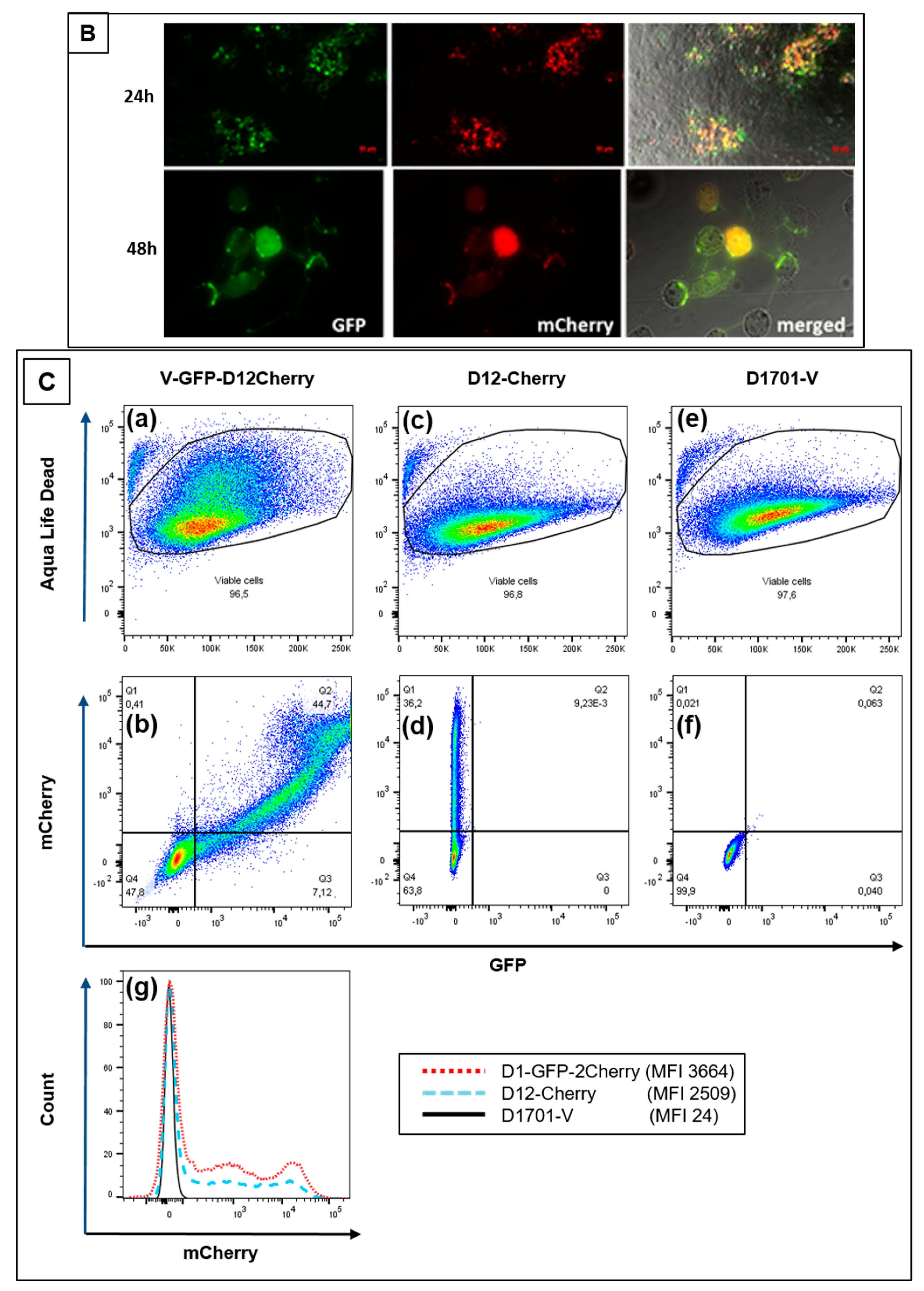
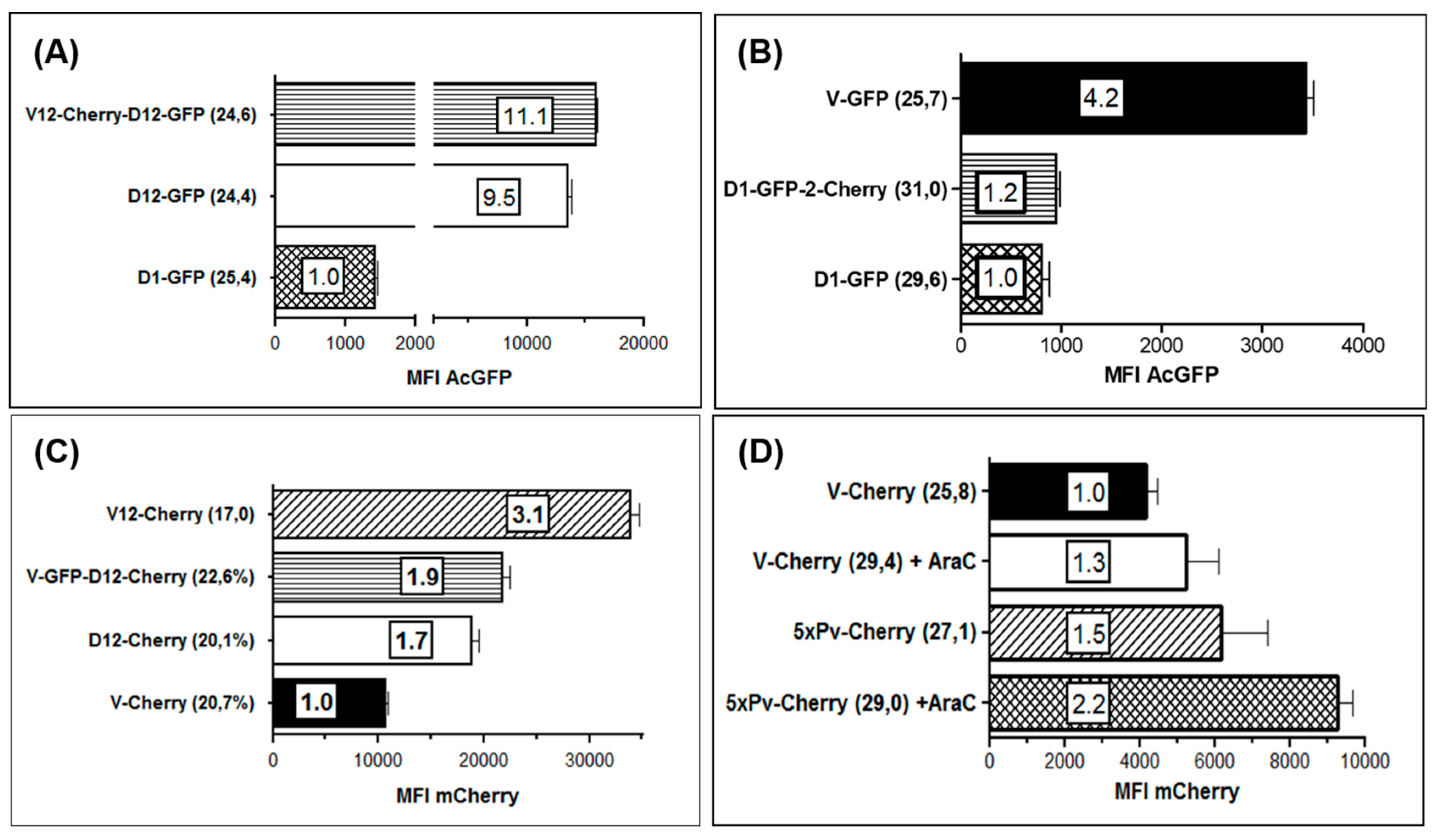
3.3. Use of Deletion D for Foreign Gene Expression
3.4. Evaluation of Expression Strength Dictated by the used ORFV Early Promoters
4. Discussion
5. Conclusions and Future Perspectives
Patent
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chan, W.M.; McFadden, G. Oncolytic Poxviruses. Annu. Rev. Virol. 2014, 1, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Draper, S.J.; Cottingham, M.G.; Gilbert, S.C. Utilizing poxviral vectored vaccines for antibody induction-progress and prospects. Vaccine 2013, 31, 4223–4230. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.E.; Najera, J.L.; Krupa, M.; Perdiguero, B.; Esteban, M. MVA and NYVAC as vaccines against emergent infectious diseases and cancer. Curr. Gene Ther. 2011, 11, 189–217. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.R.; Dolin, R. Vaccinia viruses: Vaccines against smallpox and vectors against infectious diseases and tumors. Expert Rev. Vaccines 2011, 10, 1221–1240. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.L.; Langland, J.O.; Kibler, K.V.; Denzler, K.L.; White, S.D.; Holechek, S.A.; Wong, S.; Huynh, T.; Baskin, C.R. Vaccinia virus vaccines: Past, present and future. Antivir. Res. 2009, 84, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastoret, P.P.; Vanderplasschen, A. Poxviruses as vaccine vectors. Comp. Immunol. Microbiol. Infect. Dis. 2003, 26, 343–355. [Google Scholar] [CrossRef]
- Sanchez-Sampedro, L.; Perdiguero, B.; Mejias-Perez, E.; Garcia-Arriaza, J.; Di Pilato, M.; Esteban, M. The evolution of poxvirus vaccines. Viruses 2015, 7, 1726–1803. [Google Scholar] [CrossRef]
- Gomez, C.E.; Najera, J.L.; Krupa, M.; Esteban, M. The poxvirus vectors MVA and NYVAC as gene delivery systems for vaccination against infectious diseases and cancer. Curr. Gene Ther. 2008, 8, 97–120. [Google Scholar] [CrossRef]
- Drexler, I.; Staib, C.; Sutter, G. Modified vaccinia virus Ankara as antigen delivery system: How can we best use its potential? Curr. Opin. Biotechnol. 2004, 15, 506–512. [Google Scholar] [CrossRef]
- Volz, A.; Sutter, G. Protective efficacy of Modified Vaccinia virus Ankara in preclinical studies. Vaccine 2013, 31, 4235–4240. [Google Scholar] [CrossRef]
- Gilbert, S.C. Clinical development of Modified Vaccinia virus Ankara vaccines. Vaccine 2013, 31, 4241–4246. [Google Scholar] [CrossRef] [PubMed]
- Albarnaz, J.D.; Torres, A.A.; Smith, G.L. Modulating Vaccinia Virus Immunomodulators to Improve Immunological Memory. Viruses 2018, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arriaza, J.; Esteban, M. Enhancing poxvirus vectors vaccine immunogenicity. Hum. Vaccines Immunother. 2014, 10, 2235–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büttner, M.; Rziha, H.J. Parapoxviruses: From the lesion to the viral genome. J. Vet. Med. B Infect. Dis. Vet. Public Health 2002, 49, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.B.; Wise, L.M.; Mercer, A.A. Molecular Genetic Analysis of Orf Virus: A Poxvirus That Has Adapted to Skin. Viruses 2015, 7, 1505–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haig, D.; Mercer, A.A. Parapoxviruses. In Encyclopedia of Virology, 3rd ed.; Mahy, B.W.J., Regenmortel, M.H.V.V., Eds.; Elsevier: Oxford, UK, 2008; Volume 4, pp. 57–63. [Google Scholar]
- Hussain, K.A.; Burger, D. In vivo and in vitro characteristics of contagious ecthyma virus isolates: Host response mechanism. Vet. Microbiol. 1989, 19, 23–36. [Google Scholar] [CrossRef]
- Rziha, H.J.; Henkel, M.; Cottone, R.; Bauer, B.; Auge, U.; Götz, F.; Pfaff, E.; Röttgen, M.; Dehio, C.; Büttner, M. Generation of recombinant parapoxviruses: Non-essential genes suitable for insertion and expression of foreign genes. J. Biotechnol. 2000, 83, 137–145. [Google Scholar] [CrossRef]
- Buddle, B.M.; Dellers, R.W.; Schurig, G.G. Contagious ecthyma virus-vaccination failures. Am. J. Vet. Res. 1984, 45, 263–266. [Google Scholar]
- Haig, D.M.; Mercer, A.A. Ovine diseases. Orf. Vet. Res. 1998, 29, 311–326. [Google Scholar]
- Mayr, A.; Herlyn, M.; Mahnel, H.; Danco, A.; Zach, A.; Bostedt, H. Control of contagious ecthyma in sheep by means of a new parenteral cell culture derived live vaccine. J. Med. Vet. B 1981, 28, 535–549. [Google Scholar]
- Haig, D.M.; Thomson, J.; McInnes, C.J.; Deane, D.L.; Anderson, I.E.; McCaughan, C.A.; Imlach, W.; Mercer, A.A.; Howard, C.J.; Fleming, S.B. A comparison of the anti-inflammatory and immuno-stimulatory activities of orf virus and ovine interleukin-10. Virus Res. 2002, 90, 303–316. [Google Scholar] [CrossRef]
- Siegemund, S.; Hartl, A.; von Buttlar, H.; Dautel, F.; Raue, R.; Freudenberg, M.A.; Fejer, G.; Buttner, M.; Kohler, G.; Kirschning, C.J.; et al. Conventional bone marrow-derived dendritic cells contribute to toll-like receptor-independent production of alpha/beta interferon in response to inactivated parapoxvirus ovis. J. Virol. 2009, 83, 9411–9422. [Google Scholar] [CrossRef] [PubMed]
- Weber, O.; Siegling, A.; Friebe, A.; Limmer, A.; Schlapp, T.; Knolle, P.; Mercer, A.; Schaller, H.; Volk, H.D. Inactivated parapoxvirus ovis (Orf virus) has antiviral activity against hepatitis B virus and herpes simplex virus. J. Gen. Virol. 2003, 84, 1843–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, K.; Block, A.; Ferk, G.; Beer, B.; Vollmar, A.; Lutz, H. Treatment of feline leukemia virus (FeLV) infection. Vet. Microbiol. 1999, 69, 111–113. [Google Scholar] [CrossRef]
- Kyriakis, S.C.; Tzika, E.D.; Lyras, D.N.; Tsinas, A.C.; Saoulidis, K.; Sarris, K. Effect of an inactivated parapoxvirus based immunomodulator (Baypamun) on post weaning diarrhoea syndrome and wasting pig syndrome of piglets. Res. Vet. Sci. 1998, 64, 187–190. [Google Scholar] [CrossRef]
- Weber, O.; Mercer, A.A.; Friebe, A.; Knolle, P.; Volk, H.D. Therapeutic immunomodulation using a virus-the potential of inactivated orf virus. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 32, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Cottone, R.; Büttner, M.; Bauer, B.; Henkel, M.; Hettich, E.; Rziha, H.J. Analysis of genomic rearrangement and subsequent gene deletion of the attenuated Orf virus strain D1701. Virus Res. 1998, 56, 53–67. [Google Scholar] [CrossRef]
- Pohlscheidt, M.; Langer, U.; Minuth, T.; Bodeker, B.; Apeler, H.; Horlein, H.D.; Paulsen, D.; Rubsamen-Waigmann, H.; Henzler, H.J.; Reichl, U. Development and optimisation of a procedure for the production of Parapoxvirus ovis by large-scale microcarrier cell culture in a non-animal, non-human and non-plant-derived medium. Vaccine 2008, 26, 1552–1565. [Google Scholar] [CrossRef]
- Rziha, H.J.; Henkel, M.; Cottone, R.; Meyer, M.; Dehio, C.; Büttner, M. Parapoxviruses: Potential alternative vectors for directing the immune response in permissive and non-permissive hosts. J. Biotechnol. 1999, 73, 235–242. [Google Scholar] [CrossRef]
- Rziha, H.J.; Rohde, J.; Amann, R. Generation and Selection of Orf Virus (ORFV) Recombinants. Methods Mol. Biol. 2016, 1349, 177–200. [Google Scholar] [CrossRef]
- Meyer, M.; Clauss, M.; Lepple-Wienhues, A.; Waltenberger, J.; Augustin, H.G.; Ziche, M.; Lanz, C.; Büttner, M.; Rziha, H.J.; Dehio, C. A novel vascular endothelial growth factor encoded by Orf virus, VEGF-E, mediates angiogenesis via signalling through VEGFR-2 (KDR) but not VEGFR-1 (Flt-1) receptor tyrosine kinases. EMBO J. 1999, 18, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savory, L.J.; Stacker, S.A.; Fleming, S.B.; Niven, B.E.; Mercer, A.A. Viral vascular endothelial growth factor plays a critical role in orf virus infection. J. Virol. 2000, 74, 10699–10706. [Google Scholar] [CrossRef] [PubMed]
- Wise, L.M.; Veikkola, T.; Mercer, A.A.; Savory, L.J.; Fleming, S.B.; Caesar, C.; Vitali, A.; Makinen, T.; Alitalo, K.; Stacker, S.A. Vascular endothelial growth factor (VEGF)-like protein from orf virus NZ2 binds to VEGFR2 and neuropilin-1. Proc. Natl. Acad. Sci. USA 1999, 96, 3071–3076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amann, R.; Rohde, J.; Wulle, U.; Conlee, D.; Raue, R.; Martinon, O.; Rziha, H.J. A new rabies vaccine based on a recombinant Orf virus (Parapoxvirus) expressing the rabies virus glycoprotein. J. Virol. 2012, 87, 1618–1630. [Google Scholar] [CrossRef]
- Dory, D.; Fischer, T.; Beven, V.; Cariolet, R.; Rziha, H.J.; Jestin, A. Prime-boost immunization using DNA vaccine and recombinant Orf virus protects pigs against Pseudorabies virus (Herpes suid 1). Vaccine 2006, 24, 6256–6263. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Planz, O.; Stitz, L.; Rziha, H.J. Novel recombinant parapoxvirus vectors induce protective humoral and cellular immunity against lethal herpesvirus challenge infection in mice. J. Virol. 2003, 77, 9312–9323. [Google Scholar] [CrossRef] [PubMed]
- Henkel, M.; Planz, O.; Fischer, T.; Stitz, L.; Rziha, H.J. Prevention of virus persistence and protection against immunopathology after Borna disease virus infection of the brain by a novel Orf virus recombinant. J. Virol. 2005, 79, 314–325. [Google Scholar] [CrossRef]
- Rohde, J.; Amann, R.; Rziha, H.J. New Orf Virus (Parapoxvirus) Recombinant Expressing H5 Hemagglutinin Protects Mice against H5N1 and H1N1 Influenza A Virus. PLoS ONE 2013, 8, e83802. [Google Scholar] [CrossRef]
- Rohde, J.; Schirrmeier, H.; Granzow, H.; Rziha, H.J. A new recombinant Orf virus (ORFV, Parapoxvirus) protects rabbits against lethal infection with rabbit hemorrhagic disease virus (RHDV). Vaccine 2011, 29, 9256–9264. [Google Scholar] [CrossRef]
- Van Rooij, E.M.; Rijsewijk, F.A.; Moonen-Leusen, H.W.; Bianchi, A.T.; Rziha, H.J. Comparison of different prime-boost regimes with DNA and recombinant Orf virus based vaccines expressing glycoprotein D of pseudorabies virus in pigs. Vaccine 2010, 28, 1808–1813. [Google Scholar] [CrossRef]
- Voigt, H.; Merant, C.; Wienhold, D.; Braun, A.; Hutet, E.; Le Potier, M.F.; Saalmuller, A.; Pfaff, E.; Buttner, M. Efficient priming against classical swine fever with a safe glycoprotein E2 expressing Orf virus recombinant (ORFV VrV-E2). Vaccine 2007, 25, 5915–5926. [Google Scholar] [CrossRef] [PubMed]
- Assarsson, E.; Greenbaum, J.A.; Sundstrom, M.; Schaffer, L.; Hammond, J.A.; Pasquetto, V.; Oseroff, C.; Hendrickson, R.C.; Lefkowitz, E.J.; Tscharke, D.C.; et al. Kinetic analysis of a complete poxvirus transcriptome reveals an immediate-early class of genes. Proc. Natl. Acad. Sci. USA 2008, 105, 2140–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Bruno, D.P.; Martens, C.A.; Porcella, S.F.; Moss, B. Simultaneous high-resolution analysis of vaccinia virus and host cell transcriptomes by deep RNA sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 11513–11518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Maruri-Avidal, L.; Sisler, J.; Stuart, C.A.; Moss, B. Cascade regulation of vaccinia virus gene expression is modulated by multistage promoters. Virology 2013, 447, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Reynolds, S.E.; Martens, C.A.; Bruno, D.P.; Porcella, S.F.; Moss, B. Expression profiling of the intermediate and late stages of poxvirus replication. J. Virol. 2011, 85, 9899–9908. [Google Scholar] [CrossRef] [PubMed]
- Rziha, H.-J.; Amman Interfaculty Institute for Cell Biology, Department of Immunology, Auf der Morgenstelle 15, Tübingen 72076, Germany. Unpublished work. 2015.
- Smith, G.L.; Mackett, M.; Moss, B. Recombinant vaccinia viruses as new live vaccines. Biotechnol. Genet. Eng. Rev. 1984, 2, 383–407. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.; Peng, Y.; Hao, W.; Duan, C.; Rock, D.L.; Luo, S. Generation of recombinant Orf virus using an enhanced green fluorescent protein reporter gene as a selectable marker. BMC Vet. Res. 2011, 7, 80. [Google Scholar] [CrossRef]
- Popov, S.; Mirshahidi, S.; Essono, S.; Song, R.; Wang, X.; Ruprecht, R.M. Generation of recombinant vaccinia viruses via green fluorescent protein selection. DNA Cell Biol. 2009, 28, 103–108. [Google Scholar] [CrossRef]
- Di Lullo, G.; Soprana, E.; Panigada, M.; Palini, A.; Erfle, V.; Staib, C.; Sutter, G.; Siccardi, A.G. Marker gene swapping facilitates recombinant Modified Vaccinia Virus Ankara production by host-range selection. J. Virol. Methods 2009, 156, 37–43. [Google Scholar] [CrossRef]
- Di Lullo, G.; Soprana, E.; Panigada, M.; Palini, A.; Agresti, A.; Comunian, C.; Milani, A.; Capua, I.; Erfle, V.; Siccardi, A.G. The combination of marker gene swapping and fluorescence-activated cell sorting improves the efficiency of recombinant modified vaccinia virus Ankara vaccine production for human use. J. Virol. Methods 2010, 163, 195–204. [Google Scholar] [CrossRef]
- Spehner, D.; De Carlo, S.; Drillien, R.; Weiland, F.; Mildner, K.; Hanau, D.; Rziha, H.J. Appearance of the bona fide spiral tubule of ORF virus is dependent on an intact 10-kilodalton viral protein. J. Virol. 2004, 78, 8085–8093. [Google Scholar] [CrossRef] [PubMed]
- Rziha, H.J.; Bauer, B.; Adam, K.H.; Rottgen, M.; Cottone, R.; Henkel, M.; Dehio, C.; Buttner, M. Relatedness and heterogeneity at the near-terminal end of the genome of a parapoxvirus bovis 1 strain (B177) compared with parapoxvirus ovis (Orf virus). J. Gen. Virol. 2003, 84, 1111–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, S.; Sisler, J.R.; Moss, B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. Biotechniques 1997, 23, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Moss, B. Structure of vaccinia virus early promoters. J. Mol. Biol. 1989, 210, 749–769. [Google Scholar] [CrossRef]
- Wennier, S.T.; Brinkmann, K.; Steinhausser, C.; Maylander, N.; Mnich, C.; Wielert, U.; Dirmeier, U.; Hausmann, J.; Chaplin, P.; Steigerwald, R. A novel naturally occurring tandem promoter in modified vaccinia virus ankara drives very early gene expression and potent immune responses. PLoS ONE 2013, 8, e73511. [Google Scholar] [CrossRef] [PubMed]
- Ink, B.S.; Pickup, D.J. Transcription of a poxvirus early gene is regulated both by a short promoter element and by a transcriptional termination signal controlling transcriptional interference. J. Virol. 1989, 63, 4632–4644. [Google Scholar] [PubMed]
- Czerny, C.-P.; Waldmann, R.; Scheubeck, T. Identification of three distinct antigenic sites in parapoxviruses. Arch. Virol. 1997, 142, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Cottone, R.; Buttner, M.; McInnes, C.J.; Wood, A.R.; Rziha, H.J. Orf virus encodes a functional dUTPase gene. J. Gen. Virol. 2002, 83, 1043–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delhon, G.; Tulman, E.R.; Afonso, C.L.; Lu, Z.; Concha-Bermejillo, A.; Lehmkuhl, H.D.; Piccone, M.E.; Kutish, G.F.; Rock, D.L. Genomes of the Parapoxviruses Orf Virus and Bovine Papular Stomatitis Virus. J. Virol. 2004, 78, 168–177. [Google Scholar] [CrossRef]
- Deane, D.; McInnes, C.J.; Percival, A.; Wood, A.; Thomson, J.; Lear, A.; Gilray, J.; Fleming, S.; Mercer, A.; Haig, D. Orf virus encodes a novel secreted protein inhibitor of granulocyte-macrophage colony-stimulating factor and interleukin-2. J. Virol. 2000, 74, 1313–1320. [Google Scholar] [CrossRef]
- McInnes, C.J.; Deane, D.; Haig, D.; Percival, A.; Thomson, J.; Wood, A.R. Glycosylation, disulfide bond formation and the presence of a WSXWS-like motif in the orf virus GIF protein are critical for maintaining the integrity of Binding to ovine granulocyte-macrophage colony-stimulating factor and interleukin-2. J. Virol. 2005, 79, 11205–11213. [Google Scholar] [CrossRef] [PubMed]
- McGuire, M.J.; Johnston, S.A.; Sykes, K.F. Novel immune-modulator identified by a rapid, functional screen of the Parapoxvirus ovis (Orf virus) genome. Proteome Sci. 2012, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Friederichs, S.; Krebs, S.; Blum, H.; Wolf, E.; Lang, H.; von Buttlar, H.; Buttner, M. Comparative and retrospective molecular analysis of Parapoxvirus (PPV) isolates. Virus Res. 2014, 181, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Baur, K.; Brinkmann, K.; Schweneker, M.; Patzold, J.; Meisinger-Henschel, C.; Hermann, J.; Steigerwald, R.; Chaplin, P.; Suter, M.; Hausmann, J. Immediate-early expression of a recombinant antigen by modified vaccinia virus ankara breaks the immunodominance of strong vector-specific B8R antigen in acute and memory CD8 T-cell responses. J. Virol. 2010, 84, 8743–8752. [Google Scholar] [CrossRef] [PubMed]
- Paillot, R. A systematic review of the immune-modulators Parapoxvirus ovis and Propionibacterium acnes for the prevention of respiratory disease and other infections in the horse. Vet. Immunol. Immunopathol. 2013, 153, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mercer, A.A.; Fleming, S.B.; Ueda, N. F-Box-Like Domains are Present in Most Poxvirus Ankyrin Repeat Proteins. Virus Genes 2005, 31, 127–133. [Google Scholar] [CrossRef]
- Sonnberg, S.; Fleming, S.B.; Mercer, A.A. A truncated two-alpha-helix F-box present in poxvirus ankyrin-repeat proteins is sufficient for binding the SCF1 ubiquitin ligase complex. J. Gen. Virol. 2009, 90, 1224–1228. [Google Scholar] [CrossRef]
- Sullivan, J.T.; Fleming, S.B.; Robinson, A.J.; Mercer, A.A. Sequence and transcriptional analysis of a near-terminal region of the orf virus genome. Virus Genes 1995, 11, 21–29. [Google Scholar] [CrossRef]
- Chi, X.; Zeng, X.; Li, W.; Hao, W.; Li, M.; Huang, X.; Huang, Y.; Rock, D.L.; Luo, S.; Wang, S. Genome analysis of orf virus isolates from goats in the Fujian Province of southern China. Front. Microbiol. 2015, 6, 1135. [Google Scholar] [CrossRef]
- Fleming, S.B.; Lyttle, D.J.; Sullivan, J.T.; Mercer, A.A.; Robinson, A.J. Genomic analysis of a transposition-deletion variant of orf virus reveals a 3.3 kbp region of non-essential DNA. J. Gen. Virol. 1995, 76, 2669–2978. [Google Scholar] [CrossRef]
- McInnes, C.J.; Wood, A.R.; Nettleton, P.E.; Gilray, J.A. Genomic comparison of an avirulent strain of Orf virus with that of a virulent wild type isolate reveals that the Orf virus G2L gene is non-essential for replication. Virus Genes 2001, 22, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Y.; Fabrizio, J.A.; Wilkins, S.E.; Dave, K.A.; Gorman, J.J.; Gleadle, J.M.; Fleming, S.B.; Peet, D.J.; Mercer, A.A. Ankyrin Repeat Proteins of Orf Virus Influence the Cellular Hypoxia Response Pathway. J. Virol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, J.V.; Guerra, S.; Arakawa, Y.; Dodding, M.P.; Esteban, M.; Way, M. F11-mediated inhibition of RhoA signalling enhances the spread of vaccinia virus in vitro and in vivo in an intranasal mouse model of infection. PLoS ONE 2009, 4, e8506. [Google Scholar] [CrossRef] [PubMed]
- Marsland, B.J.; Tisdall, D.J.; Heath, D.D.; Mercer, A.A. Construction of a recombinant orf virus that expresses an Echinococcus granulosus vaccine antigen from a novel genomic insertion site. Arch. Virol. 2003, 148, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Hautaniemi, M.; Ueda, N.; Tuimala, J.; Mercer, A.A.; Lahdenpera, J.; McInnes, C.J. The genome of Pseudocowpoxvirus: Comparison of a reindeer isolate and a reference strain. J. Gen. Virol. 2010, 91, 1560–1576. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Tulman, E.R.; Diel, D.G.; Khatiwada, S.; Sims, W.; Edwards, J.F.; Wen, X.; Kutish, G.F.; Rock, D.L.; Delhon, G. Coinfection with multiple strains of bovine papular stomatitis virus. Arch. Virol. 2015, 160, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Mercer, A.A.; Ueda, N.; Friederichs, S.M.; Hofmann, K.; Fraser, K.M.; Bateman, T.; Fleming, S.B. Comparative analysis of genome sequences of three isolates of Orf virus reveals unexpected sequence variation. Virus Res. 2005, 116, 146–158. [Google Scholar] [CrossRef]
- Hautaniemi, M.; Vaccari, F.; Scacliarini, A.; Laaksonen, S.; Huovilainen, A.; McInnes, C.J. Analysis of deletion within the reindeer pseudocowpoxvirus genome. Virus Res. 2011, 160, 326–332. [Google Scholar] [CrossRef]
- Li, W.; Hao, W.; Peng, Y.; Duan, C.; Tong, C.; Song, D.; Gao, F.; Li, M.; Rock, D.L.; Luo, S. Comparative genomic sequence analysis of Chinese orf virus strain NA1/11 with other parapoxviruses. Arch. Virol. 2014, 160, 253–266. [Google Scholar] [CrossRef]
- Felix, J.; Kandiah, E.; De Munck, S.; Bloch, Y.; van Zundert, G.C.; Pauwels, K.; Dansercoer, A.; Novanska, K.; Read, R.J.; Bonvin, A.M.; et al. Structural basis of GM-CSF and IL-2 sequestration by the viral decoy receptor GIF. Nat. Commun. 2016, 7, 13228. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Liang, M.; Evans, D.H. Genomic analysis of vaccinia virus strain TianTan provides new insights into the evolution and evolutionary relationships between Orthopoxviruses. Virology 2013, 442, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, P.N.; Mundt, W.; Kistner, O.; Howard, M.K. Vero cell platform in vaccine production: Moving towards cell culture-based viral vaccines. Expert Rev. Vaccines 2009, 8, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Sutter, G.; Staib, C. Vaccinia vectors as candidate vaccines: The development of modified vaccinia virus Ankara for antigen delivery. Curr. Drug Targets Infect. Disord. 2003, 3, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.; Zheng, Z.; Hao, W.; Duan, C.; Li, W.; Wang, Y.; Li, M.; Luo, S. The N Terminus of Orf Virus-Encoded Protein 002 Inhibits Acetylation of NF-κB p65 by Preventing Ser(276) Phosphorylation. PLoS ONE 2013, 8, e58854. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.B.; Fraser, K.M.; Mercer, A.A.; Robinson, A.J. Vaccinia virus-like early transcriptional control sequences flank an early gene in orf virus. Gene 1991, 97, 207–212. [Google Scholar] [CrossRef]
- Fleming, S.B.; Fraser, K.M.; Little, D.J.; Mercer, A.A. In vivo recognition of orf virus early transcriptional promoters in a vaccinia virus recombinant. Virology 1992, 187, 464–471. [Google Scholar] [CrossRef]
- Di Pilato, M.; Mejias-Perez, E.; Gomez, C.E.; Perdiguero, B.; Sorzano, C.O.; Esteban, M. New vaccinia virus promoter as a potential candidate for future vaccines. J. Gen. Virol. 2013, 94, 2771–2776. [Google Scholar] [CrossRef] [Green Version]
- Becker, P.D.; Norder, M.; Weissmann, S.; Ljapoci, R.; Erfle, V.; Drexler, I.; Guzman, C.A. Gene Expression Driven by a Strong Viral Promoter in MVA Increases Vaccination Efficiency by Enhancing Antibody Responses and Unmasking CD8(+) T Cell Epitopes. Vaccines 2014, 2, 581–600. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Promoter eP1 | 5′-AAAAAAAAAATTGAAAAATTATTCTAAATATTGCACGG-3′ |
| Promoter eP2 | 5′-AAAAATTGAAATTCTAACTTGTGTTCTTATAAATGAT-3′ |
| Spacer SP | 5′-TTTTTATCTTGTTTTTATCCTGTCTTTTTATCAGTTTTTTA GCTAGTTAAAACATAAATAGTAAAGCTAAAAAGAGACTATATCGGCGGCTGGAGTCTTGCAACAACCAGC-3′ |
| Ch-F | 5′-GCTTCAAGGTGCACATGGAGGGC-3′ |
| Ch-R | 5′-GGTGTAGTCCTCGTTGTGGGAGG-3′ |
| GFP-F | 5′-CCACAAGTTCAGCGTGAGCGGCGAG-3′ |
| GFP-R | 5′-GCGCTTCTCGTTGGGGTCCTTGGAC-3′ |
| Del2-F | 5′-GCAACAAGGTCTGCGTGCCTGCCGACC-3′ |
| Del2-R2 | 5′-CGGCGATCTGGATGGTTGGAGCCATGCC-3′ |
| pdV-F | 5′-GGTGACGGTGCTCAGCGTGGTGGCGGTTTC-3′ |
| pdV-R | 5′-CTAGCGGCGTCTTCTGGGCGGCCTTGTGGT-3′ |
| Open reading frame (ORF) | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 101 | 102 | 103 | 114 | 115 | 116 | 117 | 118 | |||||||||||||||||
| ORFV strain | Length | Identity a) | Length | Identity | Length | Identity | Length | Identity | Length | Identity | Length | Identity | Length | Identity | Length | Identity | ||||||||
| nt | aa | nt | nt | aa | nt | nt | aa | nt | nt | aa | nt | nt | aa | nt | nt | aa | nt | nt | aa | nt | nt | aa | nt | |
| D1701-V | 3483 | 1161 | 100% | ― | ― | ― | ― | ― | ― | ― | ― | ― | ― | ― | ― | ― | ― | ― | ― | ― | ― | 306 | 102 | 100% |
| D1701-B | 3483 | 1161 | 100% | 1557 | 519 | 100% | 1548 | 516 | 100% | 1038 | 346 | 100% | 447 | 148 | 100% | 672 | 224 | 100% | 795 | 265 | 100% | 306 | 102 | 100% |
| D1701-McG | 3483 | 1161 | 99% | 1557 | 519 | 100% | 1548 | 516 | 99% | 1038 | 346 | 99% | 447 | 148 | 100% | 672 | 224 | 99% | 912 | 304 | 99% | 306 | 102 | 100% |
| NZ2 | 3483 | 1161 | 99% | 1560 | 520 | 95% | 1548 | 516 | 98% | 1038 | 346 | 96% | 435 | 145 | 88% | 693 | 231 | 81% | 795 | 265 | 96% | 357 | 119 | 97% |
| OV-IA82 | 3483 | 1161 | 99% | 1556 | 518 | 85% | 1566 | 522 | 65% | 1038 | 346 | 96% | 429 | 143 | 89% | 702 | 234 | 81% | 795 | 265 | 96% | 663 | 221 | 97% |
| B029 | 3483 | 1161 | 99% | 1560 | 520 | 95% | ― | ― | ― | 1038 | 346 | 97% | 435 | 145 | 89% | 669 | 223 | 83% | 795 | 265 | 96% | ― | ― | ― |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rziha, H.-J.; Büttner, M.; Müller, M.; Salomon, F.; Reguzova, A.; Laible, D.; Amann, R. Genomic Characterization of Orf Virus Strain D1701-V (Parapoxvirus) and Development of Novel Sites for Multiple Transgene Expression. Viruses 2019, 11, 127. https://doi.org/10.3390/v11020127
Rziha H-J, Büttner M, Müller M, Salomon F, Reguzova A, Laible D, Amann R. Genomic Characterization of Orf Virus Strain D1701-V (Parapoxvirus) and Development of Novel Sites for Multiple Transgene Expression. Viruses. 2019; 11(2):127. https://doi.org/10.3390/v11020127
Chicago/Turabian StyleRziha, Hanns-Joachim, Mathias Büttner, Melanie Müller, Ferdinand Salomon, Alena Reguzova, Dominic Laible, and Ralf Amann. 2019. "Genomic Characterization of Orf Virus Strain D1701-V (Parapoxvirus) and Development of Novel Sites for Multiple Transgene Expression" Viruses 11, no. 2: 127. https://doi.org/10.3390/v11020127
APA StyleRziha, H.-J., Büttner, M., Müller, M., Salomon, F., Reguzova, A., Laible, D., & Amann, R. (2019). Genomic Characterization of Orf Virus Strain D1701-V (Parapoxvirus) and Development of Novel Sites for Multiple Transgene Expression. Viruses, 11(2), 127. https://doi.org/10.3390/v11020127