Viral Diversity of Microbats within the South West Botanical Province of Western Australia


Abstract
1. Introduction
2. Materials and Methods
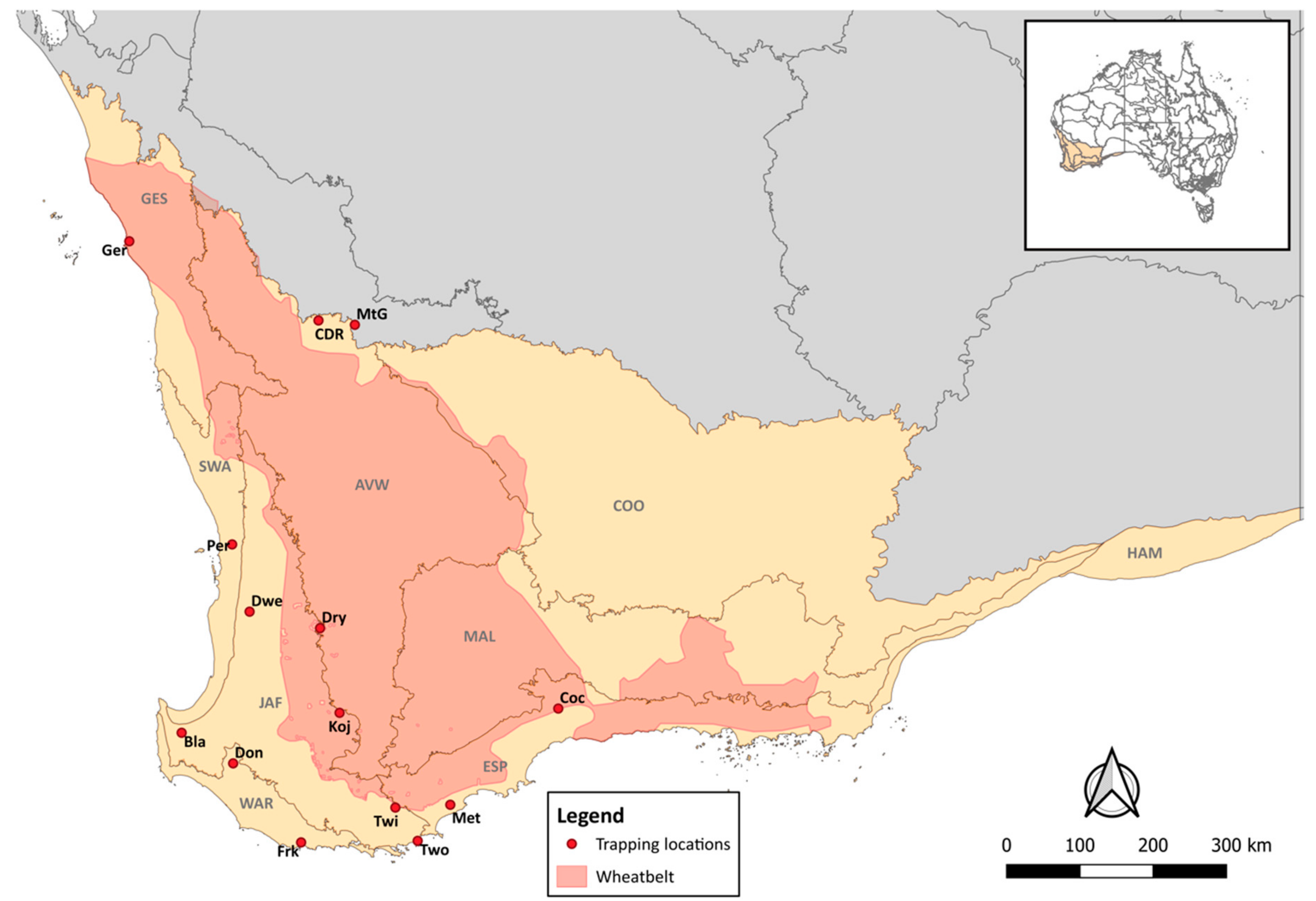
2.1. Study Area
2.2. Sample Collection
2.3. Molecular Analysis
2.4. Phylogenetic Analysis
2.5. Serology
2.6. Prevalence Estimates
3. Results
3.1. Serology
3.2. Faecal PCR Analysis
3.3. Summary of Virus Prevalence in Relation to Serology
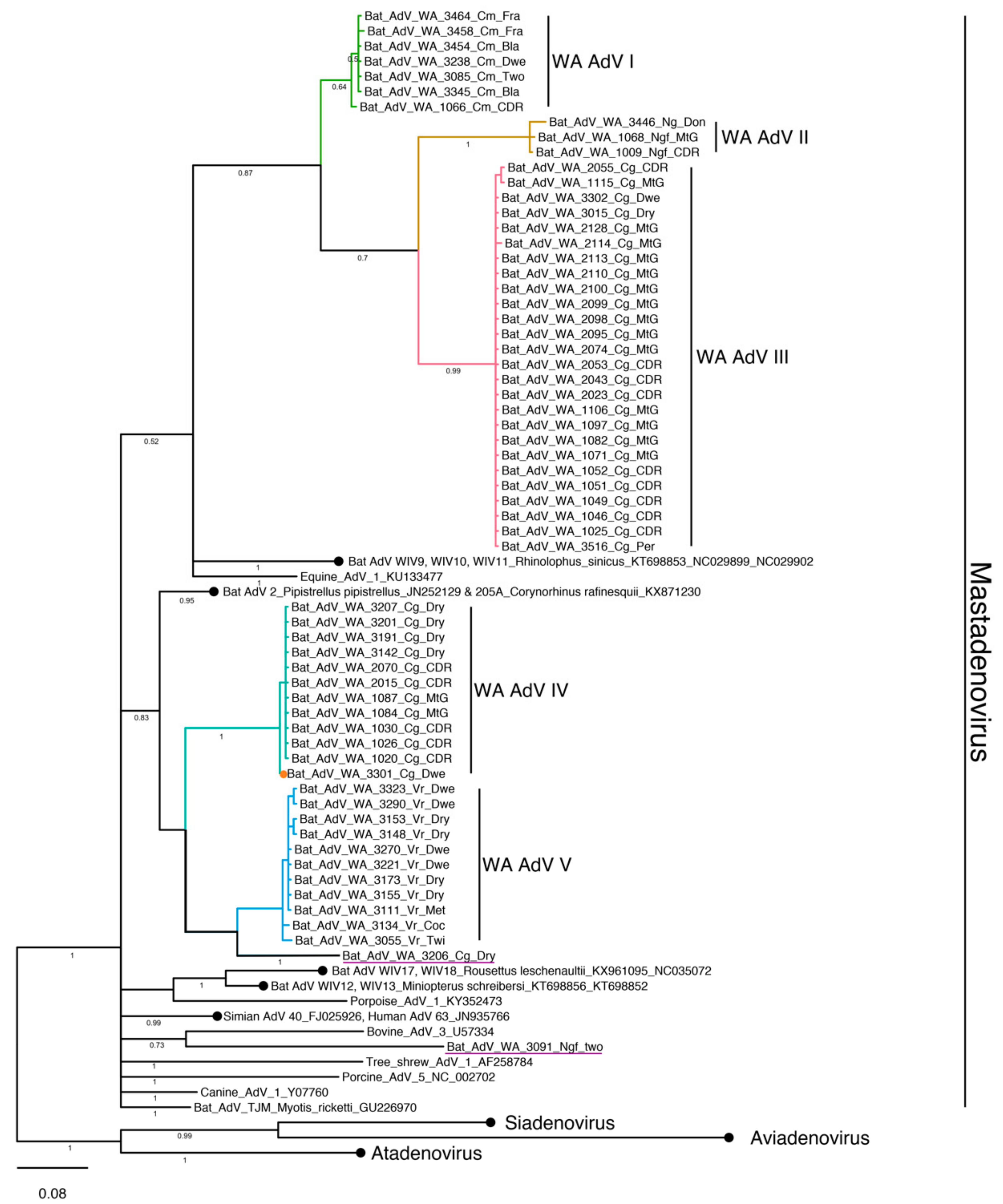
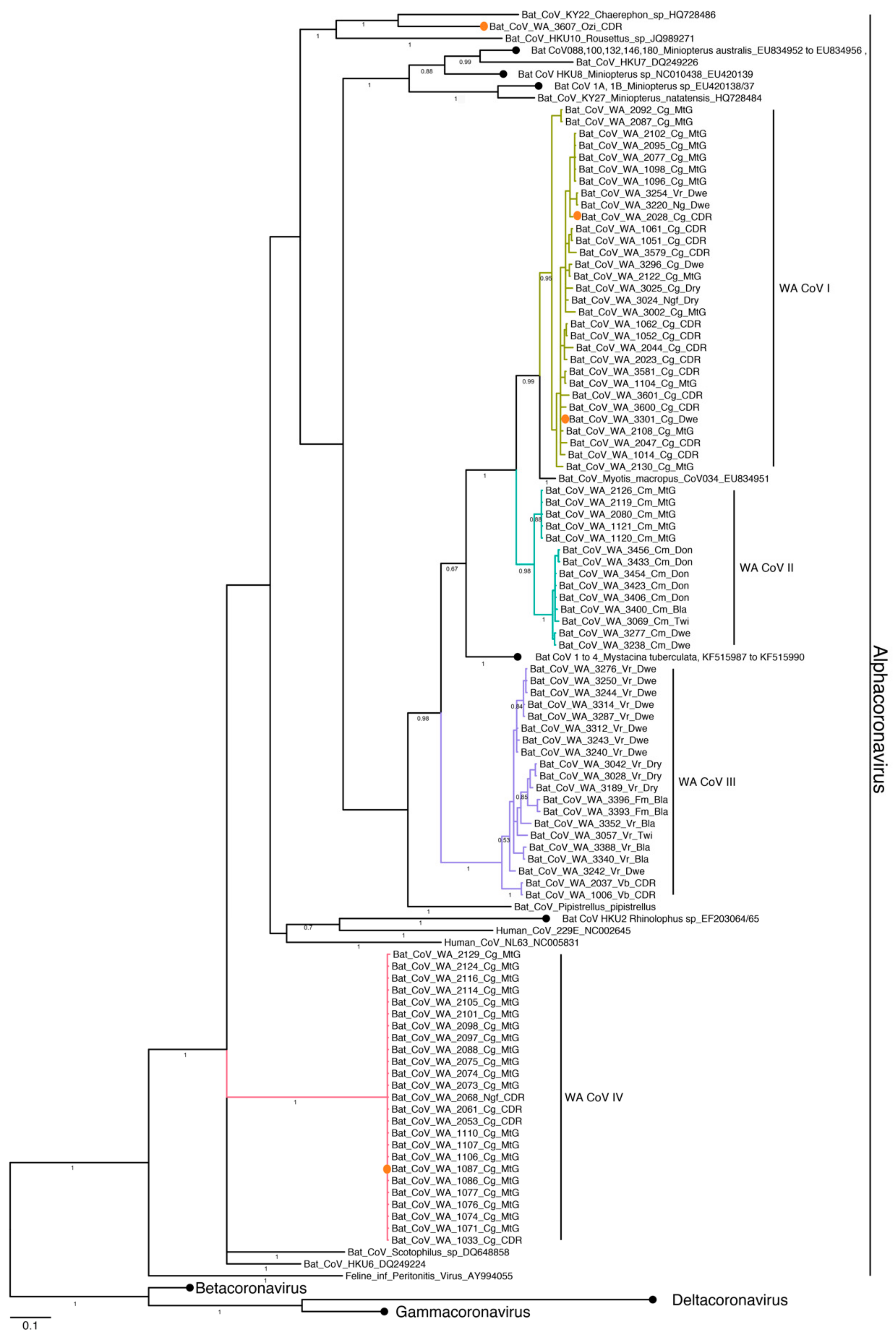
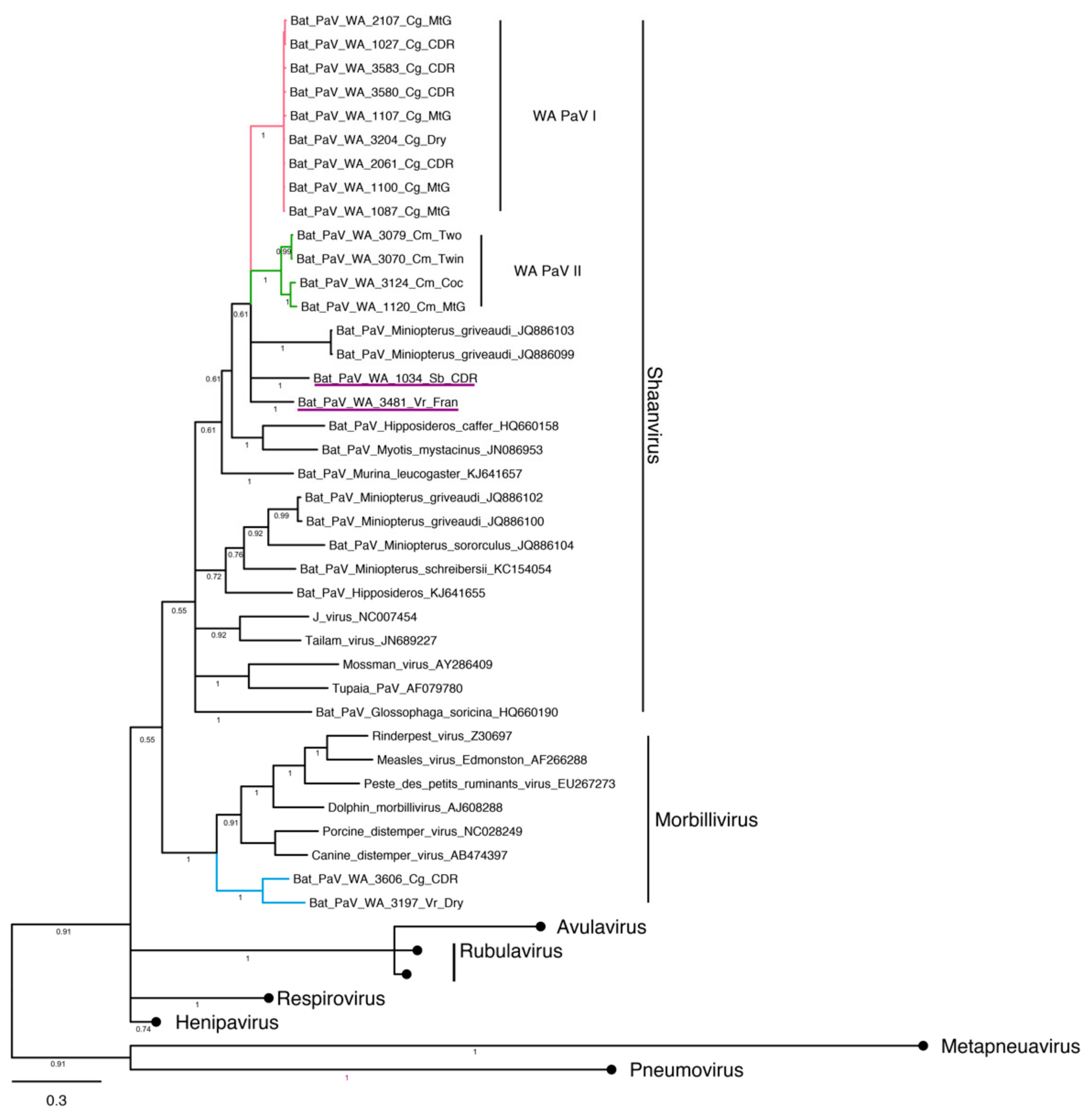
3.4. Phylogenetic Analysis and Whole Genome Sequencing
3.4.1. Adenovirus
3.4.2. Coronavirus
3.4.3. Paramyxovirus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Drexler, J.F.; Corman, V.M.; Drosten, C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res. 2014, 101, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Marsh, G.A.; de Jong, C.; Barr, J.A.; Tachedjian, M.; Smith, C.; Middleton, D.; Yu, M.; Todd, S.; Foord, A.J.; Haring, V.; et al. Cedar Virus: A novel Henipavirus isolated from Australian bats. PLoS Pathog. 2012, 8, e1002836. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D.; Daszak, P.; Wolfe, N.D.; Gao, G.F.; Morel, C.M.; Morzaria, S.; Pablos-Méndez, A.; Tomori, O.; Mazet, J.A.K. The Global Virome Project. Science (80-. ) 2018, 359, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Bradley, C.A.; Altizer, S. Urbanization and the ecology of wildlife diseases. Trends Ecol. Evol. 2007, 22, 95–102. [Google Scholar] [CrossRef]
- Altizer, S.; Ostfeld, R.S.; Johnson, P.T.J.; Kutz, S.; Harvell, C.D. Climate change and infectious diseases: from evidence to a predictive framework. Science (80-. ) 2013, 341, 514–519. [Google Scholar] [CrossRef]
- Plowright, R.K.; Foley, P.; Field, H.E.; Dobson, A.P.; Foley, J.E.; Eby, P.; Daszak, P. Urban habituation, ecological connectivity and epidemic dampening: the emergence of Hendra virus from flying foxes (Pteropus spp.). Proceedings. Biol. Sci. 2011, 278, 3703–3712. [Google Scholar] [CrossRef]
- Walsh, M.G.; Wiethoelter, A.; Haseeb, M.A. The impact of human population pressure on flying fox niches and the potential consequences for Hendra virus spillover. Sci. Rep. 2017, 7, 8226. [Google Scholar] [CrossRef]
- Jung, K.; Threlfall, C.G. Urbanisation and Its Effects on Bats—A Global Meta-Analysis. In Bats in the Anthropocene: Conservation of Bats in a Changing World; Voigt, C.C., Kingston, T., Eds.; Springer International Publishing: London, UK, 2016; pp. 13–33. [Google Scholar]
- Roberts, K.E.; Hadfield, J.D.; Sharma, M.D.; Longdon, B. Changes in temperature alter the potential outcomes of virus host shifts. PLOS Pathog. 2018, 14, e1007185. [Google Scholar] [CrossRef]
- Gervasi, S.S.; Burgan, S.C.; Hofmeister, E.; Unnasch, T.R.; Martin, L.B. Stress hormones predict a host superspreader phenotype in the West Nile virus system. Proc. R. Soc. B Biol. Sci. 2017, 284, 20171090. [Google Scholar] [CrossRef]
- Davy, C.M.; Donaldson, M.E.; Subudhi, S.; Rapin, N.; Warnecke, L.; Turner, J.M.; Bollinger, T.K.; Kyle, C.J.; Dorville, N.A.S.-Y.; Kunkel, E.L.; et al. White-nose syndrome is associated with increased replication of a naturally persisting coronaviruses in bats. Sci. Rep. 2018, 8, 15508. [Google Scholar] [CrossRef]
- Giles, J.R.; Eby, P.; Parry, H.; Peel, A.J.; Plowright, R.K.; Westcott, D.A.; McCallum, H. Environmental drivers of spatiotemporal foraging intensity in fruit bats and implications for Hendra virus ecology. Sci. Rep. 2018, 8, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, D.M.; Carver, S.; Jones, M.E.; Krkošek, M.; Skerratt, L.F. Emerging infectious diseases of wildlife: A critical perspective. Trends Parasitol. 2015, 31, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Hayman, D.T.S. Bats as viral reservoirs. Annu. Rev. Virol. 2016, 3, 77–99. [Google Scholar] [CrossRef] [PubMed]
- Memish, Z.A.; Mishra, N.; Olival, K.J.; Fagbo, S.F.; Kapoor, V.; Epstein, J.H.; AlHakeem, R.; Durosinloun, A.; Al Asmari, M.; Islam, A.; et al. Middle East Respiratory Syndrome Coronavirus in bats, Saudi Arabia. Emerg. Infect. Dis. 2013, 19, 1819–1824. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Woo, P.C.Y.; Li, K.S.M.; Huang, Y.; Tsoi, H.-W.; Wong, B.H.L.; Wong, S.S.Y.; Leung, S.-Y.; Chan, K.-H.; Yuen, K.-Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef]
- Reusken, C.B.; Haagmans, B.L.; Müller, M.A.; Gutierrez, C.; Godeke, G.-J.; Meyer, B.; Muth, D.; Raj, V.S.; Vries, L.S.-D.; Corman, V.M.; et al. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect. Dis. 2013, 13, 859–866. [Google Scholar] [CrossRef]
- Li, Y.; Ge, X.; Zhang, H.; Zhou, P.; Zhu, Y.; Zhang, Y.; Yuan, J.; Wang, L.-F.; Shi, Z. Host range, prevalence, and genetic diversity of adenoviruses in bats. J. Virol. 2010, 84, 3889–3897. [Google Scholar] [CrossRef]
- Chen, E.C.; Yagi, S.; Kelly, K.R.; Mendoza, S.P.; Maninger, N.; Rosenthal, A.; Spinner, A.; Bales, K.L.; Schnurr, D.P.; Lerche, N.W.; et al. Cross-species transmission of a novel adenovirus associated with a fulminant pneumonia outbreak in a new world monkey colony. PLoS Pathog. 2011, 7, e1002155. [Google Scholar] [CrossRef]
- Mahalingam, S.; Herrero, L.J.; Playford, E.G.; Spann, K.; Herring, B.; Rolph, M.S.; Middleton, D.; McCall, B.; Field, H.; Wang, L.-F. Hendra virus: an emerging paramyxovirus in Australia. Lancet Infect. Dis. 2012, 12, 799–807. [Google Scholar] [CrossRef]
- Mohd Nor, M.N.; Gan, C.H.; Ong, B.L. Nipah virus infection of pigs in peninsular Malaysia. Rev. Sci. Tech. 2000, 19, 160–165. [Google Scholar] [CrossRef]
- Field, H.; Jordan, D.; Edson, D.; Morris, S.; Melville, D.; Parry-Jones, K.; Broos, A.; Divljan, A.; McMichael, L.; Davis, R.; et al. Spatiotemporal aspects of hendra virus infection in pteropid bats (Flying-Foxes) in Eastern Australia. PLoS ONE 2015, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.S.; Field, H.E.; Guyatt, K.J. Managing emerging diseases borne by fruit bats (flying foxes), with particular reference to henipaviruses and Australian bat lyssavirus. J. Appl. Microbiol. 2003, 94, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.H.J.; Baker, M.L. Bats and bat-borne diseases: a perspective on Australian megabats. Aust. J. Zool. 2013, 61, 48. [Google Scholar] [CrossRef]
- Gould, A.R.; Kattenbelt, J.A.; Gumley, S.G.; Lunt, R.A. Characterisation of an Australian bat lyssavirus variant isolated from an insectivorous bat. Virus Res. 2002, 89, 1–28. [Google Scholar] [CrossRef]
- Field, H.E. Evidence of Australian bat lyssavirus infection in diverse Australian bat taxa. Zoonoses Public Health 2018, 65, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Prada, D.; Boyd, V.; Baker, M.; Jackson, B.; O’Dea, M. Insights into Australian Bat Lyssavirus in insectivorous bats of Western Australia. Trop. Med. Infect. Dis. 2019, 4, 46. [Google Scholar] [CrossRef]
- Smith, C.S.; De Jong, C.E.; Meers, J.; Henning, J.; Wang, L.-F.; Field, H.E. Coronavirus infection and diversity in bats in the Australasian region. Ecohealth 2016, 13, 72–82. [Google Scholar] [CrossRef]
- Jeong, J.; Smith, C.S.; Peel, A.J.; Plowright, R.K.; Kerlin, D.H.; McBroom, J.; McCallum, H. Persistent infections support maintenance of a coronavirus in a population of Australian bats (Myotis macropus). Epidemiol. Infect. 2017, 145, 2053–2061. [Google Scholar] [CrossRef]
- Holz, P.H.; Lumsden, L.F.; Druce, J.; Legione, A.R.; Vaz, P.; Devlin, J.M.; Hufschmid, J. Virus survey in populations of two subspecies of bent-winged bats (Miniopterus orianae bassanii and oceanensis) in south-eastern Australia reveals a high prevalence of diverse herpesviruses. PLoS ONE 2018, 13, e0197625. [Google Scholar] [CrossRef]
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; da Fonseca, G.A.; Kent, J. Biodiversity hotspots for conservation priorities. Nature 2000, 403, 853–858. [Google Scholar] [CrossRef]
- Wardell-Johnson, G.; Wardell-Johnson, A.; Bradby, K.; Robinson, T.; Bateman, P.W.; Williams, K.; Keesing, A.; Braun, K.; Beckerling, J.; Burbridge, M. Application of a Gondwanan perspective to restore ecological integrity in the south-western Australian global biodiversity hotspot. Restor. Ecol. 2016, 1–11. [Google Scholar] [CrossRef]
- Churchill, S. Australian bats, 2nd ed.; Allen & Unwin: Crows Nest, Australia, 2008. [Google Scholar]
- Goldstein, T. (University of California, Davis, CA, USA). Personal communication, 2017.
- Tong, S.; Chern, S.-W.W.; Li, Y.; Pallansch, M.A.; Anderson, L.J. Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. J. Clin. Microbiol. 2008, 46, 2652–2658. [Google Scholar] [CrossRef] [PubMed]
- O’Dea, M.A.; Jackson, B.; Jackson, C.; Xavier, P.; Warren, K. Discovery and partial genomic characterisation of a novel nidovirus associated with respiratory disease in wild shingleback lizards (Tiliqua rugosa). PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Vandenberghe, L.H.; Kryazhimskiy, S.; Grant, R.; Calcedo, R.; Yuan, X.; Keough, M.; Sandhu, A.; Wang, Q.; Medina-Jaszek, C.A.; et al. Isolation and characterization of adenoviruses persistently shed from the gastrointestinal tract of non-human primates. PLoS Pathog. 2009, 5, e1000503. [Google Scholar] [CrossRef]
- Lacroix, A.; Duong, V.; Hul, V.; San, S.; Davun, H.; Omaliss, K.; Chea, S.; Hassanin, A.; Theppangna, W.; Silithammavong, S.; et al. Genetic diversity of coronaviruses in bats in Lao PDR and Cambodia. Infect. Genet. Evol. 2017, 48, 10–18. [Google Scholar] [CrossRef]
- Quan, P.-L.; Firth, C.; Street, C.; Henriquez, J.A.; Petrosov, A.; Tashmukhamedova, A.; Hutchison, S.K.; Egholm, M.; Osinubi, M.O.V.; Niezgoda, M.; et al. Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. MBio 2010, 1, e00208-10. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Bossart, K.N.; McEachern, J.A.; Hickey, A.C.; Choudhry, V.; Dimitrov, D.S.; Eaton, B.T.; Wang, L.-F. Neutralization assays for differential henipavirus serology using Bio-Plex Protein Array Systems. J. Virol. Methods 2007, 142, 29–40. [Google Scholar] [CrossRef]
- Boyd, V.; Smith, I.; Crameri, G.; Burroughs, A.L.; Durr, P.a.; White, J.; Cowled, C.; Marsh, G.a.; Wang, L.F. Development of multiplexed bead arrays for the simultaneous detection of nucleic acid from multiple viruses in bat samples. J. Virol. Methods 2015, 223, 5–12. [Google Scholar] [CrossRef]
- Hayman, D.T.S.; Wang, L.F.; Barr, J.; Baker, K.S.; Suu-Ire, R.; Broder, C.C.; Cunningham, A.A.; Wood, J.L.N. Antibodies to henipavirus or henipa-like viruses in domestic pigs in Ghana, West Africa. PLoS ONE 2011, 6, e25256. [Google Scholar] [CrossRef]
- Hayman, D.T.S.; Suu-Ire, R.; Breed, A.C.; McEachern, J.A.; Wang, L.; Wood, J.L.N.; Cunningham, A.A. Evidence of henipavirus infection in West African fruit bats. PLoS ONE 2008, 3, e2739. [Google Scholar] [CrossRef]
- Plowright, R.K.; Field, H.E.; Smith, C.; Divljan, A.; Palmer, C.; Tabor, G.; Daszak, P.; Foley, J.E. Reproduction and nutritional stress are risk factors for Hendra virus infection in little red flying foxes ( Pteropus scapulatus ). Proc. R. Soc. B Biol. Sci. 2008, 275, 861–869. [Google Scholar] [CrossRef]
- Breed, A.C.; Yu, M.; Barr, J.A.; Crameri, G.; Thalmann, C.M.; Wang, L.F. Prevalence of henipavirus and rubulavirus antibodies in pteropid bats, Papua New Guinea. Emerg. Infect. Dis. 2010, 16, 1997–1999. [Google Scholar] [CrossRef]
- Brown, L.D.; Cai, T.T.; Dasgupta, A. Interval estimation for a binomial proportion. Stat. Sci. 2001, 16, 101–133. [Google Scholar] [CrossRef]
- Aragon, T. epitools: Epidemiology Tools. R package version 0.5-10. Published 26 October 2017. Available online: https://CRAN.R-project.org/package=epitools (accessed on 20 April 2019).
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Caballero, M.; Juste, J.; Vázquez-Morón, S.; Falcon, A.; Aznar-Lopez, C.; Ibáñez, C.; Pozo, F.; Ruiz, G.; Berciano, J.M.; Garin, I.; et al. New adenovirus groups in western palaearctic bats. Viruses 2018, 10, 443. [Google Scholar] [CrossRef]
- Vidovszky, M.Z.; Kohl, C.; Boldogh, S.; Görföl, T.; Wibbelt, G.; Kurth, A.; Harrach, B. Random sampling of the Central European bat fauna reveals the existence of numerous hitherto unknown adenoviruses. Acta Vet. Hung. 2015, 63, 508–525. [Google Scholar] [CrossRef]
- Wray, A.K.; Olival, K.J.; Morán, D.; Lopez, M.R.; Alvarez, D.; Navarrete-Macias, I.; Liang, E.; Simmons, N.B.; Lipkin, W.I.; Daszak, P.; et al. Viral diversity, prey preference, and Bartonella prevalence in Desmodus rotundus in Guatemala. Ecohealth 2016, 13, 761–774. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Poon, L.L.M.; Chan, K.H.; Chen, H.; Guan, Y.; Yuen, K.Y.; Peiris, J.S.M. Coronaviruses in bent-winged bats (Miniopterus spp.). J. Gen. Virol. 2006, 87, 2461–2466. [Google Scholar] [CrossRef]
- Dominguez, S.R.; O’Shea, T.J.; Oko, L.M.; Holmes, K.V. Detection of group 1 coronaviruses in bats in North America. Emerg. Infect. Dis. 2007, 13, 1295–1300. [Google Scholar] [CrossRef]
- Moreira-Soto, A.; Taylor-Castillo, L.; Vargas-Vargas, N.; Rodríguez-Herrera, B.; Jiménez, C.; Corrales-Aguilar, E. Neotropical bats from Costa Rica harbour diverse coronaviruses. Zoonoses Public Health 2015, 62, 501–505. [Google Scholar] [CrossRef]
- Wilkinson, D.A.; Temmam, S.; Lebarbenchon, C.; Lagadec, E.; Chotte, J.; Guillebaud, J.; Ramasindrazana, B.; Héraud, J.-M.; de Lamballerie, X.; Goodman, S.M.; et al. Identification of novel paramyxoviruses in insectivorous bats of the Southwest Indian Ocean. Virus Res. 2012, 170, 159–163. [Google Scholar] [CrossRef]
- Yuan, L.; Li, M.; Li, L.; Monagin, C.; Chmura, A.A.; Schneider, B.S.; Epstein, J.H.; Mei, X.; Shi, Z.; Daszak, P.; et al. Evidence for retrovirus and paramyxovirus infection of multiple bat species in china. Viruses 2014, 6, 2138–2154. [Google Scholar] [CrossRef]
- Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Göttsche, M.; Panning, M.; Drexler, J.F.; Petersen, N.; Annan, A.; Grywna, K.; Müller, M.; et al. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg. Infect. Dis. 2008, 14, 626–631. [Google Scholar] [CrossRef]
- Rizzo, F.; Edenborough, K.M.; Toffoli, R.; Culasso, P.; Zoppi, S.; Dondo, A.; Robetto, S.; Rosati, S.; Lander, A.; Kurth, A.; et al. Coronavirus and paramyxovirus in bats from Northwest Italy. BMC Vet. Res. 2017, 13, 396. [Google Scholar] [CrossRef]
- Asano, K.M.; Hora, A.S.; Scheffer, K.C.; Fahl, W.O.; Iamamoto, K.; Mori, E.; Brandão, P.E. Alphacoronavirus in urban Molossidae and Phyllostomidae bats, Brazil. Virol. J. 2016, 13, 110. [Google Scholar] [CrossRef]
- Zheng, X.Y.; Qiu, M.; Chen, H.F.; Chen, S.W.; Xiao, J.P.; Jiang, L.N.; Huo, S.T.; Shi, T.L.; Ma, L.Z.; Liu, S.; et al. Molecular detection and phylogenetic characterization of bat and human adenoviruses in Southern China. Vector-Borne Zoonotic Dis. 2016, 16, 423–427. [Google Scholar] [CrossRef]
- Conrardy, C.; Tao, Y.; Kuzmin, I.V.; Niezgoda, M.; Agwanda, B.; Breiman, R.F.; Anderson, L.J.; Rupprecht, C.E.; Tong, S. Short report: Molecular detection of adenoviruses, rhabdoviruses, and paramyxoviruses in bats from Kenya. Am. J. Trop. Med. Hyg. 2014, 91, 258–266. [Google Scholar] [CrossRef]
- Lumsden, L.F.; Bennett, A.F.; Silins, J.E. Selection of roost sites by the lesser long-eared bat (Nyctophilus geoffroyi) and Gould’s wattled bat (Chalinolobus gouldii) in south-eastern Australia. J. Zool. 2002, 257, S095283690200081X. [Google Scholar] [CrossRef]
- Rueegger, N.; Law, B.; Goldingay, R. Interspecific differences and commonalities in maternity roosting by tree cavity-roosting bats over a maternity season in a timber production landscape. PLoS ONE 2018, 13, e0194429. [Google Scholar] [CrossRef]
- Burgar, J.M.; Craig, M.D.; Stokes, V.L. The importance of mature forest as bat roosting habitat within a production landscape. For. Ecol. Manage. 2015, 356, 112–123. [Google Scholar] [CrossRef]
- Bender, R. (The Melbourne bat box monitoring program, Melbourne, Australia). Personal communication, 2018.
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.-L.; Shi, W.-F.; Zhang, W.; Zhu, Y.; Zhang, Y.-W.; Xie, Q.-M.; Mani, S.; et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–258. [Google Scholar] [CrossRef]
- Benkö, M.; Harrach, B. Molecular evolution of adenoviruses. In Adenoviruses: Model and Vectors in Virus-Host Interactions; Doerfler, W., Böhm, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2003; pp. 3–35. [Google Scholar]
- Hackenbrack, N.; Rogers, M.B.; Ashley, R.E.; Keel, M.K.; Kubiski, S.V.; Bryan, J.A.; Ghedin, E.; Holmes, E.C.; Hafenstein, S.L.; Allison, A.B. Evolution and Cryo-electron Microscopy Capsid Structure of a North American Bat Adenovirus and Its Relationship to Other Mastadenoviruses. J. Virol. 2017, 91, e01504-16. [Google Scholar] [CrossRef]
- Sonntag, M.; Mühldorfer, K.; Speck, S.; Wibbelt, G.; Kurth, A. New adenovirus in bats, Germany. Emerg. Infect. Dis. 2009, 15, 2052–2055. [Google Scholar] [CrossRef]
- Jánoska, M.; Vidovszky, M.; Molnár, V.; Liptovszky, M.; Harrach, B.; Benkő, M. Novel adenoviruses and herpesviruses detected in bats. Vet. J. 2011, 189, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Ren, X.; Yang, L.; Hu, Y.; Yang, J.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Du, J.; et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J. Virol. 2012, 86, 10999–11012. [Google Scholar] [CrossRef] [PubMed]
- Bensch, S.; Hellgren, O.; Pérez-Tris, J. MalAvi: a public database of malaria parasites and related haemosporidians in avian hosts based on mitochondrial cytochrome b lineages. Mol. Ecol. Resour. 2009, 9, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Cottontail, V.M.; Rasche, A.; Yordanov, S.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Zeus, V.; Kwasnitschka, L.; Kerth, G.; Haase, M.; Groschup, M.H.; Balkema-Buschmann, A. Insectivorous bats carry host specific astroviruses and coronaviruses across different regions in Germany. Infect. Genet. Evol. 2016, 37, 108–116. [Google Scholar] [CrossRef]
- Peel, A.J.; McKinley, T.J.; Baker, K.S.; Barr, J.A.; Crameri, G.; Hayman, D.T.S.; Feng, Y.-R.; Broder, C.C.; Wang, L.-F.; Cunningham, A.A.; et al. Use of cross-reactive serological assays for detecting novel pathogens in wildlife: Assessing an appropriate cutoff for henipavirus assays in African bats. J. Virol. Methods 2013, 193, 295–303. [Google Scholar] [CrossRef]
- Burroughs, A.L.; Durr, P.A.; Boyd, V.; Graham, K.; White, J.R.; Todd, S.; Barr, J.; Smith, I.; Baverstock, G.; Meers, J.; et al. Hendra virus infection dynamics in the grey-headed flying fox (Pteropus poliocephalus) at the southern-most extent of its range: Further evidence this species does not readily transmit the virus to horses. PLoS ONE 2016, 11, e0155252. [Google Scholar] [CrossRef]
- Wong, S.; Lau, S.; Woo, P.; Yuen, K.-Y. Bats as a continuing source of emerging infections in humans. Rev. Med. Virol. 2007, 17, 67–91. [Google Scholar] [CrossRef]
- Luis, A.D.; Hayman, D.T.S.; O’Shea, T.J.; Cryan, P.M.; Gilbert, A.T.; Pulliam, J.R.C.; Mills, J.N.; Timonin, M.E.; Willis, C.K.R.; Cunningham, A.A.; et al. A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc. Biol. Sci. 2013, 280, 1–9. [Google Scholar] [CrossRef]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef]
- Turmelle, A.S.; Olival, K.J. Correlates of Viral Richness in Bats (Order Chiroptera). Ecohealth 2009, 6, 522–539. [Google Scholar] [CrossRef] [PubMed]
- Maganga, G.D.; Bourgarel, M.; Vallo, P.; Dallo, T.D.; Ngoagouni, C.; Drexler, J.F.; Drosten, C.; Nakouné, E.R.; Leroy, E.M.; Morand, S. Bat distribution size or shape as determinant of viral richness in African bats. PLoS ONE 2014, 9, e100172. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.; Olival, K.J.; Bumrungsri, S.; Siriaroonrat, B.; Bourgarel, M.; Morand, S. Parasite and viral species richness of Southeast Asian bats: Fragmentation of area distribution matters. Int. J. Parasitol. Parasites Wildl. 2014, 3, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Bordes, F.; Morand, S. The impact of multiple infections on wild animal hosts: a review. Infect. Ecol. Epidemiol. 2011, 1. [Google Scholar] [CrossRef]
- Woolhouse, M.E.J.; Gowtage-Sequeria, S. Host range and emerging and reemerging pathogens. Emerg. Infect. Dis. 2005, 11, 1842–1847. [Google Scholar] [CrossRef]
- Streicker, D.G.; Turmelle, A.S.; Vonhof, M.J.; Kuzmin, I.V.; McCracken, G.F.; Rupprecht, C.E. Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science (80-. ) 2010, 329, 676–679. [Google Scholar] [CrossRef]
- Basham, R.; Law, B.; Banks, P. Microbats in a “leafy” urban landscape: Are they persisting, and what factors influence their presence? Austral Ecol. 2011, 36, 663–678. [Google Scholar] [CrossRef]
- Straka, T.M.; Lentini, P.E.; Lumsden, L.F.; Wintle, B.A.; van der Ree, R. Urban bat communities are affected by wetland size, quality, and pollution levels. Ecol. Evol. 2016, 6, 4761–4774. [Google Scholar] [CrossRef]
- Bullen, R.D.; McKenzie, N.L. Seasonal range variation of Tadarida australis (Chiroptera : Molossidae) in Western Australia: The impact of enthalpy. Aust. J. Zool. 2005, 53, 145–156. [Google Scholar] [CrossRef]
- Hammarin, A.L.; Berndtsson, L.T.; Falk, K.; Nedinge, M.; Olsson, G.; Lundkvist, Å. Lyssavirus-reactive antibodies in Swedish bats. Infect. Ecol. Epidemiol. 2016, 6, 31262. [Google Scholar] [CrossRef]
- Amengual, B.; Bourhy, H.; López-Roig, M.; Serra-Cobo, J. Temporal dynamics of European bat lyssavirus type 1 and survival of Myotis myotis bats in natural colonies. PLoS ONE 2007, 2, e566. [Google Scholar] [CrossRef] [PubMed]
- Peel, A.J.; Wells, K.; Giles, J.; Boyd, V.; Burroughs, A.; Edson, D.; Crameri, G.; Baker, M.L.; Field, H.; Wang, L.-F.; et al. Synchronous shedding of multiple bat paramyxoviruses coincides with peak periods of Hendra virus spillover. Emerg. Microbes Infect. 2019, 8, 1314–1323. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Group | Target Region | Primer Name | T °C | Amplicon Size | Reference |
|---|---|---|---|---|---|
| Adenovirus | DNA polymerase gene (DNApol) | F1 | 55 | 205 | [39] |
| R1 | |||||
| F2 | 55 | ||||
| R2 | |||||
| Coronavirus | RNA-dependent RNA polymerase gene (RdRp) | CoV-Fwd1 | 53 | 400 | [40] |
| CoV-Rvs2 | |||||
| CoV-Fwd2 | 50 | ||||
| PLQ-F1 | 50 | 300 | [41] | ||
| PLQ-R1 | |||||
| PLQ-F2 | 50 | ||||
| PLQ-R2 | |||||
| Paramyxovirus | RNA-dependent RNA polymerase gene (RdRp) | PAR-F1 | 53 | 500 | [35] |
| PAR-R | |||||
| PAR-F2 | 50 | ||||
| RES-MOR-HEN-F1 | 53 | 500 | |||
| RES-MOR-HEN-R | |||||
| RES-MOR-HEN-F2 | 50 |
| Family | Species | N Faecal | N Serum | Trapping Locations |
|---|---|---|---|---|
| Vespertilionidae | Chalinolobus gouldii | 232 | 264 | MtG, CDR, Coc, Dry, Dwe, Koj, Per |
| Chalinolobus morio | 45 | 66 | MtG, Bla, CDR, Coc, Don, Dry, Dwe, Frk, Met, Twi, Two | |
| Falsistrellus mackenziei | 11 | 7 | Bla, Dwe | |
| Nyctophilus geoffroyi | 51 | 50 | MtG, Bla, CDR, Coc, Don, Dry, Dwe, Ger, Per, Two | |
| Nyctophilus gouldi | 56 | 72 | Bla, Don, Dry, Dwe, Per, Two | |
| Nyctophilus major | 10 | 6 | Bla, CDR, Dry, Dwe, Frk | |
| Scotorepens balstoni | 9 | 8 | MtG, CDR | |
| Vespadelus baverstocki | 4 | 5 | MtG, CDR | |
| Vespadelus regulus | 141 | 170 | Bla, Coc, Don, Dry, Dwe, Frk Met, Per, Twi, Two | |
| Molossidae | Austronomus australis | 9 | 11 | CDR |
| Ozimops sp | 3 | 3 | CDR |
| Family | Genus | Antigen | N 1 | Pos.2 | Seroprevalence % |
|---|---|---|---|---|---|
| Paramyxoviridae | Henipavirus | HeV | 645 | 33 | 5.1 (3.6–7.1) |
| CedV | 637 | 34 | 5.3 (3.8–7.3) | ||
| NiV | 648 | 9 | 1.3 (0.7–2.6) | ||
| Rubulavirus | TioV | 653 | 17 | 2.6 (1.6–4.1) | |
| Coronaviridae | Betacoronavirus | SARS-CoV | 645 | 38 | 5.8 (4.3–7.9) |
| MERS-CoV | 659 | 10 | 1.5 (0.8–2.7) |
| Paramyxovirus Antigens | Betacoronavirus Antigens | Overall Summary | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HeV | CedV | NiV | TioV | SARS-CoV | MERS-CoV | |||||||||
| Species | N | SeP | N | SeP | N | SeP | N | SeP | N | SeP | N | SeP | PaV Range | CoV Range |
| C. gouldii | 66(2) | 3 (0.8–10.4) | 66(4) | 6 (2.4–14.6) | 68(2) | 3 (0.8–10) | 68(4) | 6 (2.3–14.2) | 66(2) | 3 (0.8–10.4) | 68(2) | 3 (0.8–10.1) | 3–6 | 3 |
| C. morio | 60(5) | 8.3 (3.6–18.0) | 58(6) | 10.3 (4.8–20.7) | 59(0) | 58(2) | 3.3 (0.9–11.5) | 59(4) | 6.7 (2.6–16) | 60(1) | 1.6 (0.2–8.7) | 3.3–10.3 | 1.6–6.7 | |
| F. mackenziei | 7(7) | NC | 7(7) | NC | 7(3) | NC | 7(0) | 7(0) | 7(0) | NC | ||||
| N. geoffroyi | 27(0) | 26(0) | 26(1) | 3.8 (0.6–1.8) | 26(0) | 26(0) | 27(0) | NC | ||||||
| N. gouldi | 69(2) | 3 (0.8–10) | 63(2) | 1.6 (0.3–8.4) | 67(0) | 69(0) | 67(7) | 10 (5.1–20) | 70(0) | 1.6–3 | 10 | |||
| N. major | 5(0) | 5(0) | 5(0) | 5(1) | NC | 5(2) | NC | 4(0) | NC | NC | ||||
| S. balstoni | 2(0) | 2(0) | 2(0) | 2(0) | 2(0) | 2(0) | ||||||||
| V. baverstocki | 1(0) | 1(0) | 1(0) | 1(0) | 1(0) | 1(0) | ||||||||
| V. regulus | 147(7) | 4.8 (2.3–9.5) | 146(9) | 6.1 (3.2–11.3) | 150(1) | 0.6 (0.1–3.7) | 154(2) | 1.3 (0.3–4.6) | 150(9) | 6 (3–11) | 155(4) | 2.5 (1.0–6.4) | 0.6–6.1 | 2.5–6 |
| A. australis | 11(0) | 11(0) | 11(0) | 11(1) | NC | 11(1) | NC | 11(0) | NC | NC | ||||
| O. sp | 1(0) | 1(0) | 1(0) | 1(0) | 1(0) | 1(0) | ||||||||
| Family | Species | N | Adenoviridae | Coronaviridae | Paramyxoviridae |
|---|---|---|---|---|---|
| Vespertilionidae | C. gouldii | 232 | 17 (13–23) (41) | 25 (20–31) (59) | 4 (2–7) (10) |
| C. morio | 45 | 16 (8–29) (7) | 36 (24–51) (17) | 9 (3–21) (4) | |
| F. mackenziei | 11 | NC1 (2) | |||
| N. geoffroyi | 51 | 4 (1–13) (2) | 4 (1–13) (2) | 2 (0.3–10) (1) | |
| N. gouldi | 56 | 1.8 (0.3–9) (1) | 3.5 (1–12) (2) | ||
| N. major | 10 | ||||
| S. balstoni | 9 | NC1 (1) | |||
| V. baverstocki | 4 | NC1 (2) | |||
| V. regulus | 141 | 8 (4–13) (11) | 12 (8–18) (17) | 1.4 (0.4–5) (2) | |
| Molossidae | A. australis | 9 | |||
| O. sp | 3 | NC1 (1) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prada, D.; Boyd, V.; Baker, M.L.; O’Dea, M.; Jackson, B. Viral Diversity of Microbats within the South West Botanical Province of Western Australia. Viruses 2019, 11, 1157. https://doi.org/10.3390/v11121157
Prada D, Boyd V, Baker ML, O’Dea M, Jackson B. Viral Diversity of Microbats within the South West Botanical Province of Western Australia. Viruses. 2019; 11(12):1157. https://doi.org/10.3390/v11121157
Chicago/Turabian StylePrada, Diana, Victoria Boyd, Michelle L. Baker, Mark O’Dea, and Bethany Jackson. 2019. "Viral Diversity of Microbats within the South West Botanical Province of Western Australia" Viruses 11, no. 12: 1157. https://doi.org/10.3390/v11121157
APA StylePrada, D., Boyd, V., Baker, M. L., O’Dea, M., & Jackson, B. (2019). Viral Diversity of Microbats within the South West Botanical Province of Western Australia. Viruses, 11(12), 1157. https://doi.org/10.3390/v11121157