Fowl Adenovirus (FAdV) Recombination with Intertypic Crossovers in Genomes of FAdV-D and FAdV-E, Displaying Hybrid Serological Phenotypes




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Strains and DNA Preparation
2.2. Sequencing and Genome Assembly
2.3. Phylogenetic Analyses and Investigation of Positive Selection Patterns
2.4. Genomic and Antigenic Composition of FAdV-D and FAdV-E Strains
2.5. Coevolution and Recombination Analyses
2.6. Cross-Neutralization Testing
3. Results
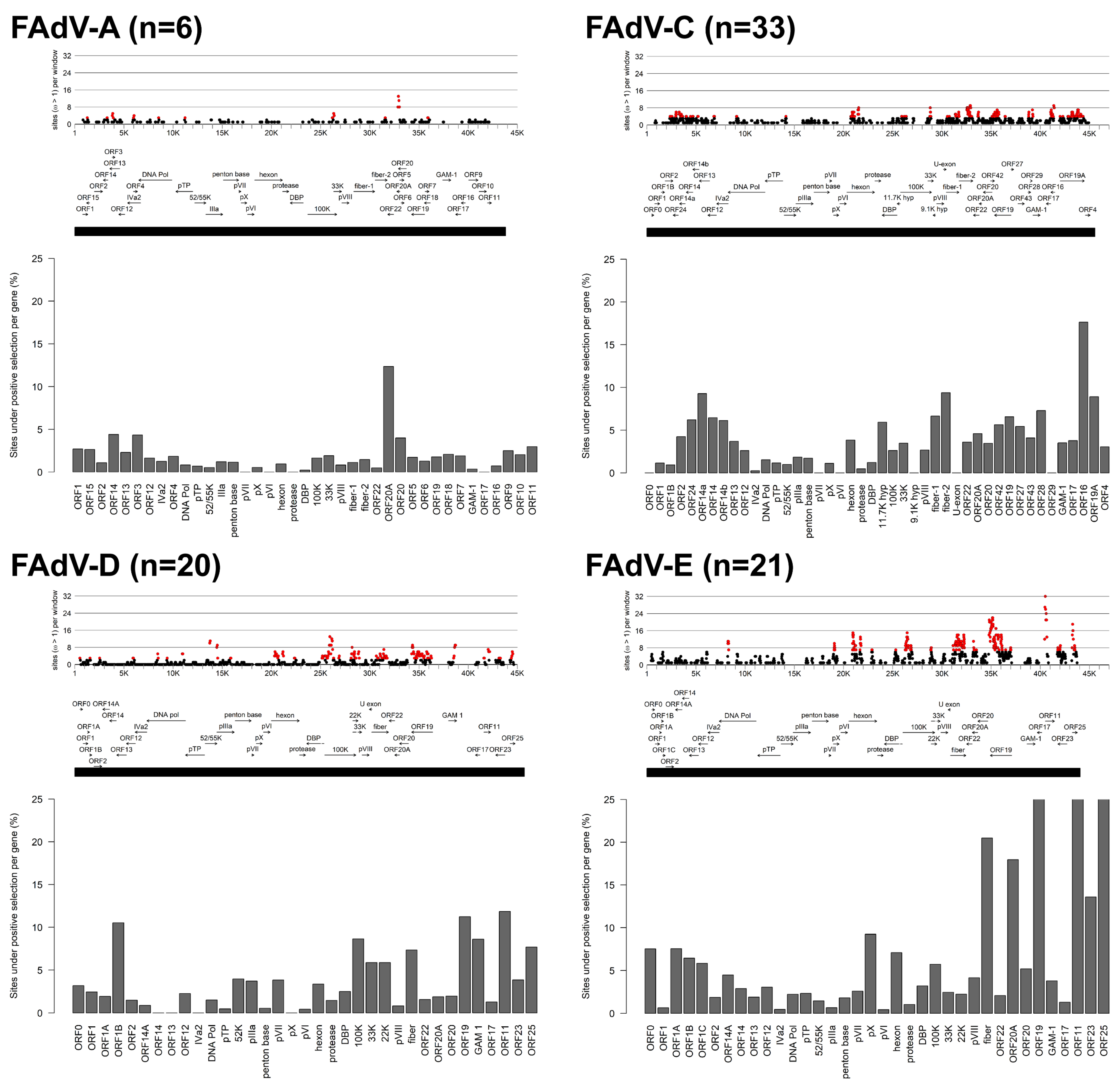
3.1. Molecular Phylogeny and Positive Selection Patterns of FAdVs
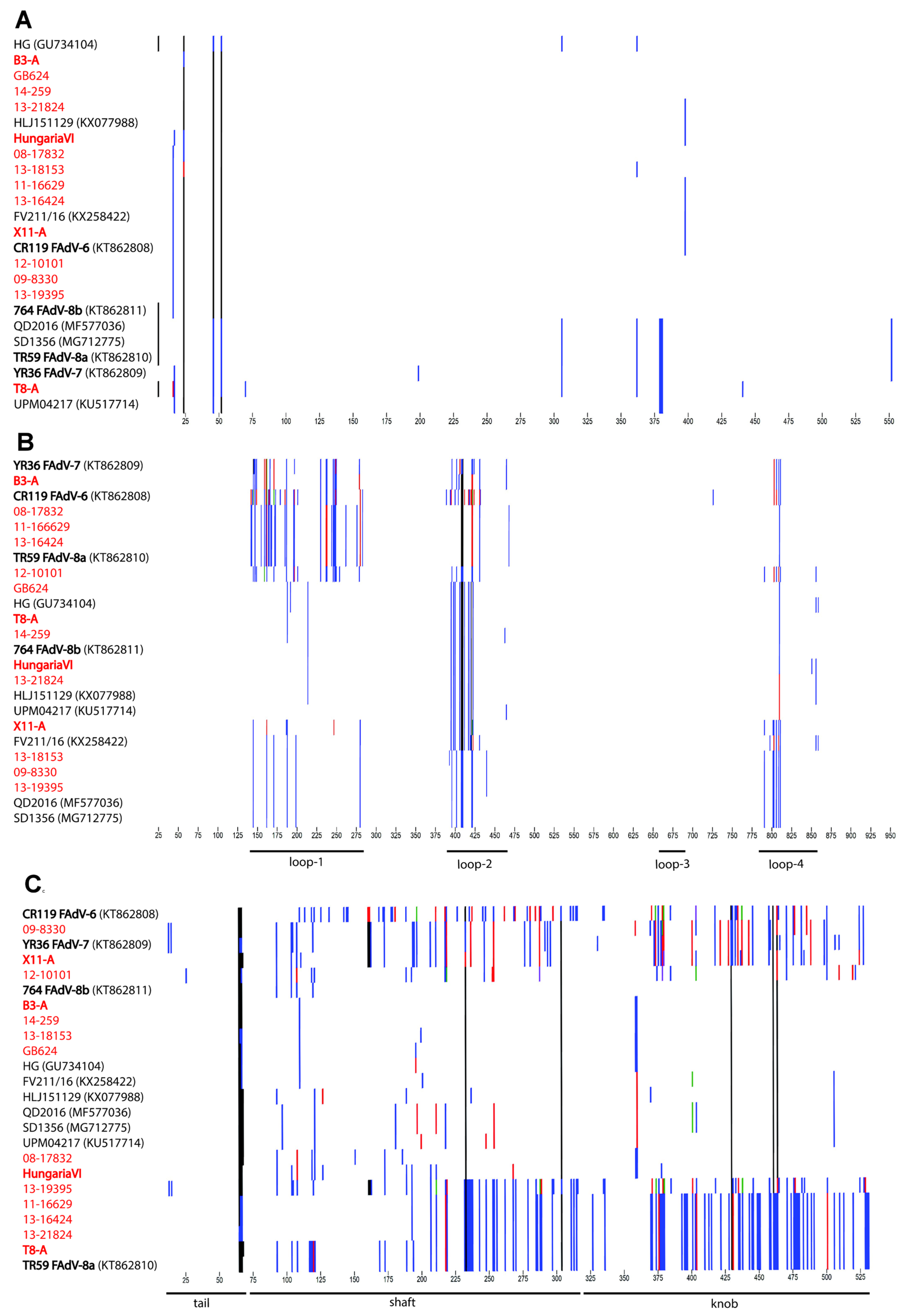
3.2. Genomic and Antigenic Composition of FAdV-D and FAdV-E Strains
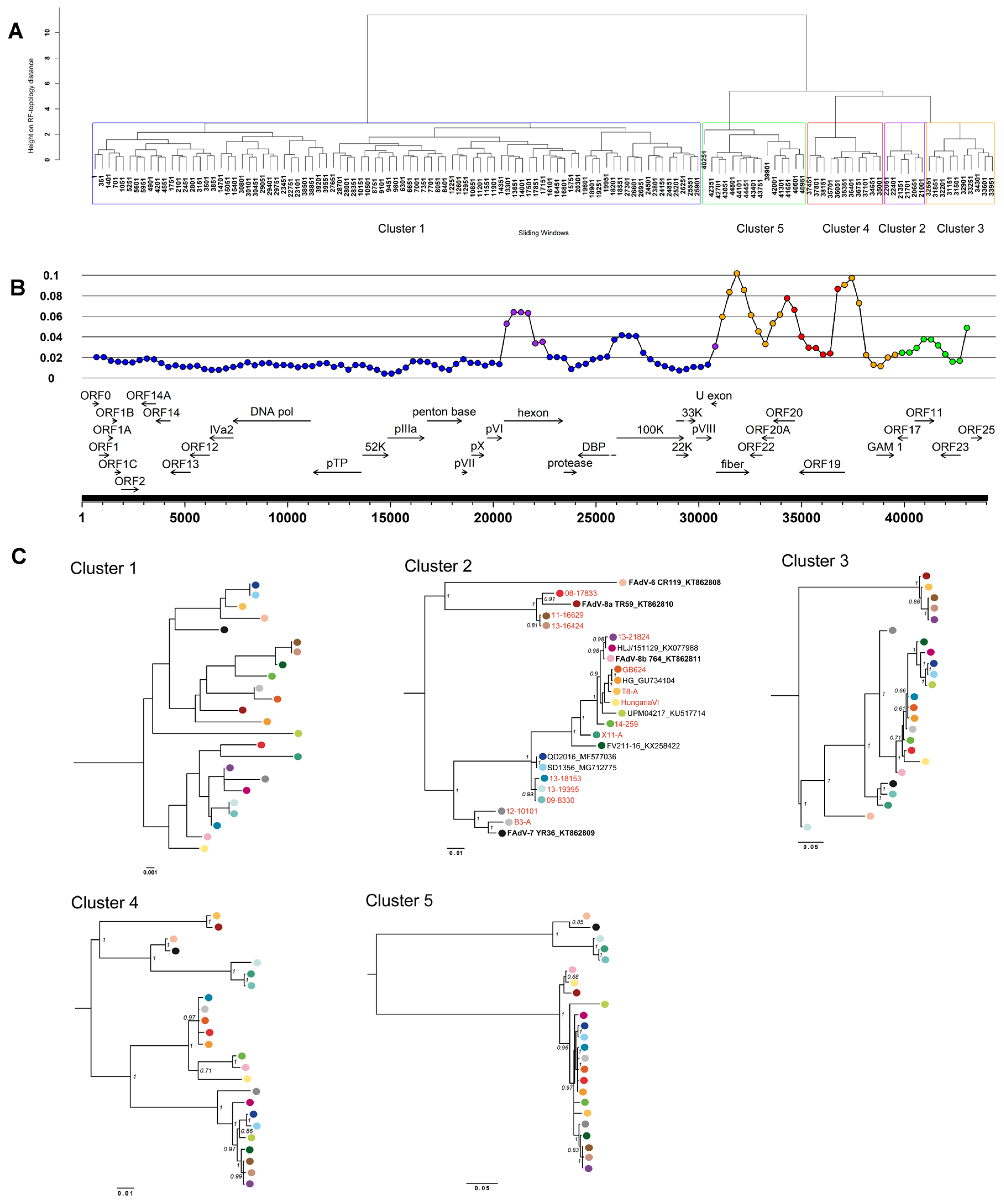
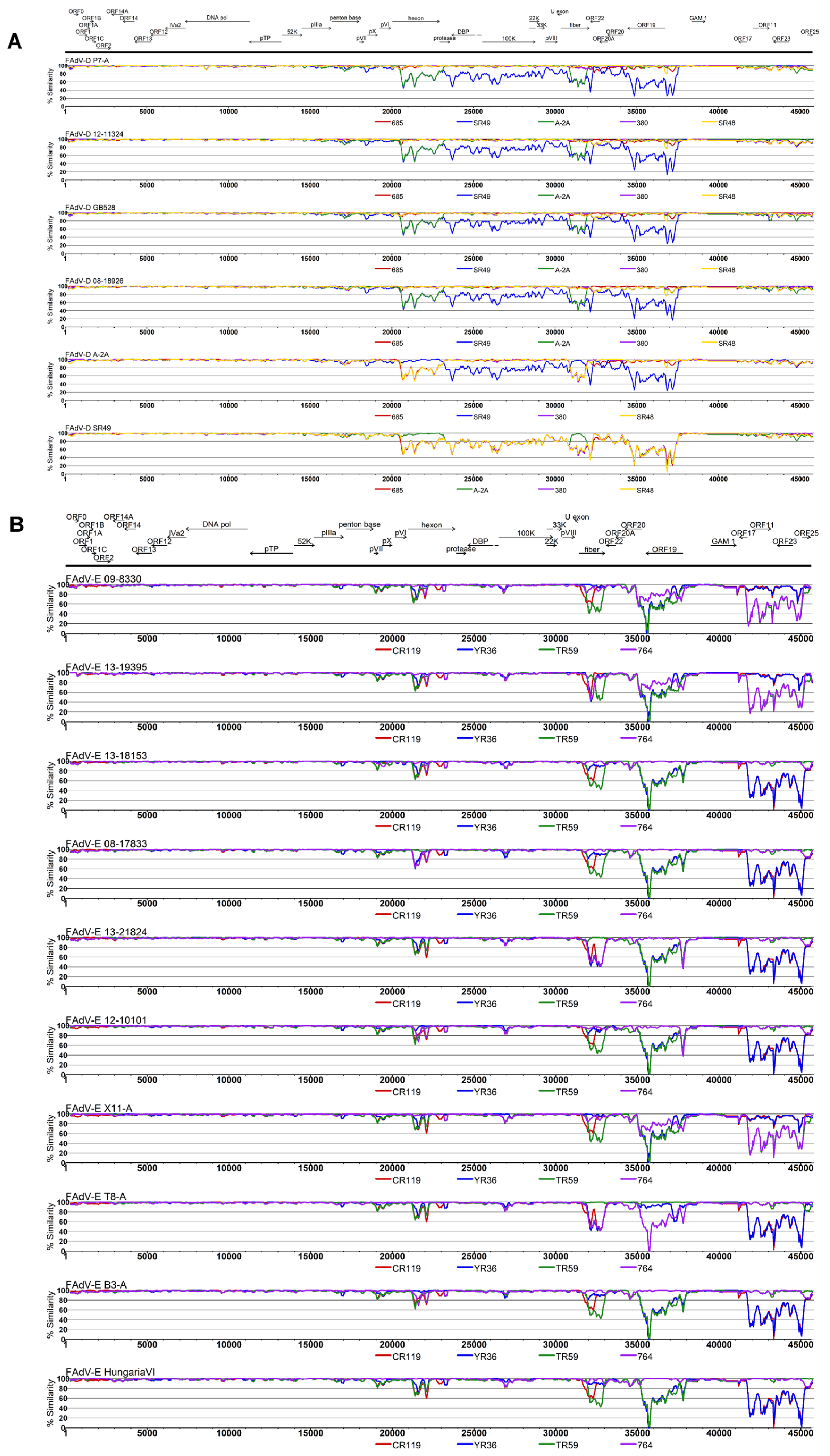
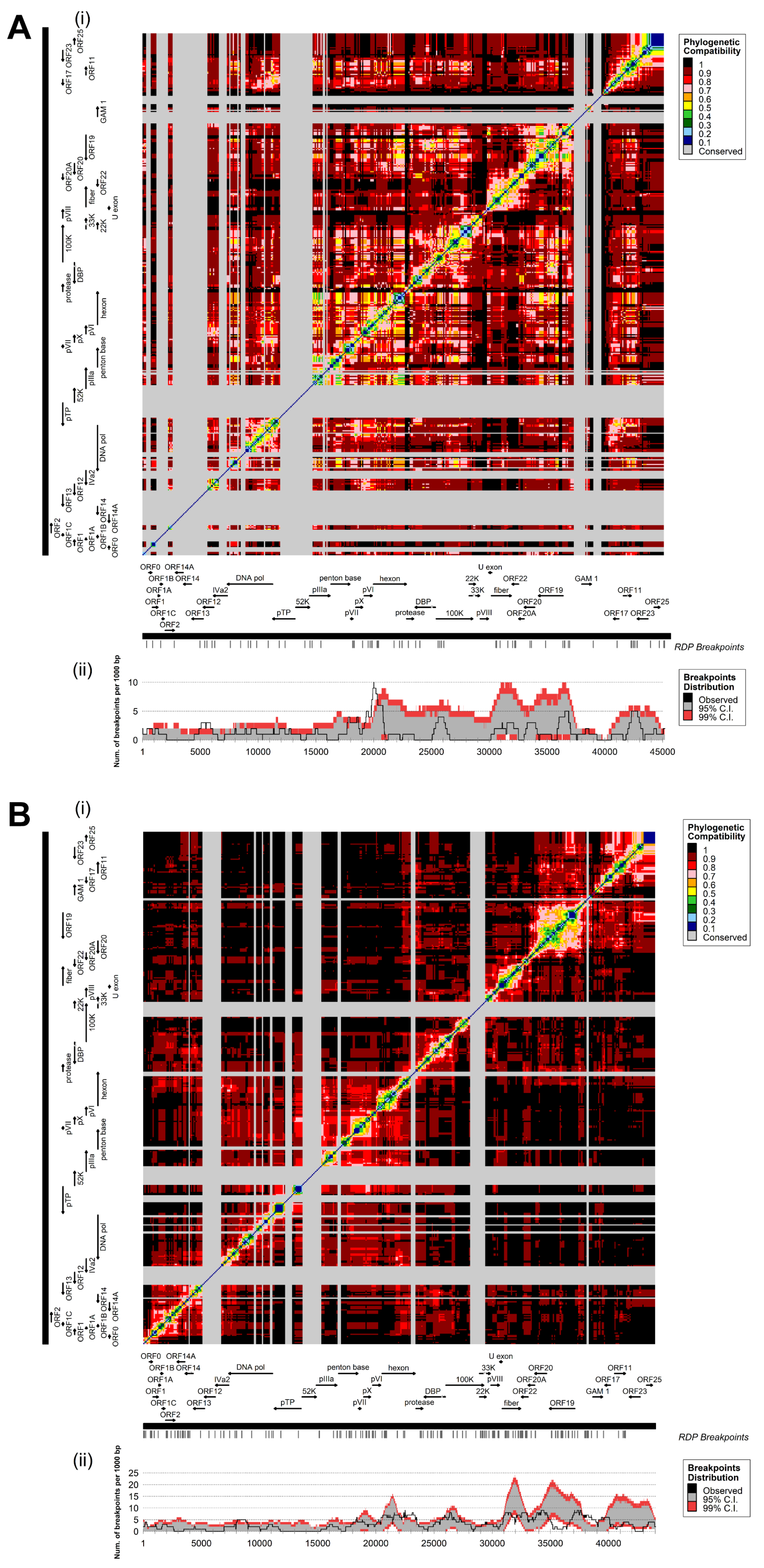
3.3. Coevolution and Recombination Analyses of FAdV-D and FAdV-E
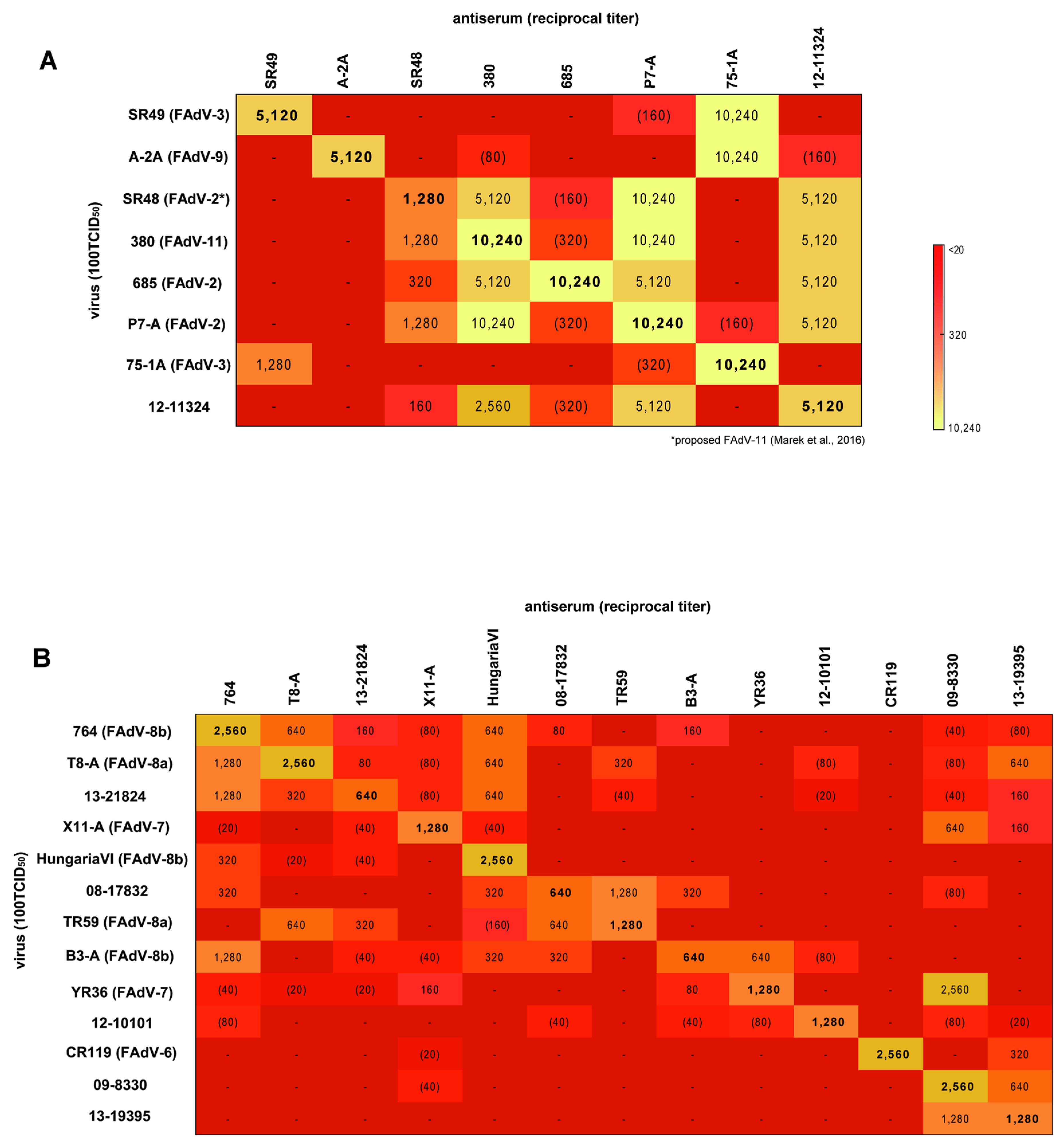
3.4. Cross-Neutralization Assays
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hess, M. Detection and differentiation of avian adenoviruses: A review. Avian Pathol. 2000, 29, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Marek, A.; Gunes, A.; Schulz, E.; Hess, M. Classification of fowl adenoviruses by use of phylogenetic analysis and high-resolution melting-curve analysis of the hexon L1 gene region. J. Virol. Methods 2010, 170, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Harrach, B.; Benkö, M.; Both, G.W.; Brown, M.; Davison, A.J.; Echavarría, M.; Hess, M.; Jones, M.S.; Kajon, A.; Lehmkuhl, H.D.; et al. Family Adenoviridae. In Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; the International Committee on Taxonomy of Viruses, King, A.M.Q., Adams, M.J., Lefkowitz, E.J., Carstens, E.B., Eds.; Academic Press: Cambridge, MA, USA, 2011; pp. 125–141. [Google Scholar]
- Raue, R.; Hess, M. Hexon based PCRs combined with restriction enzyme analysis for rapid detection and differentiation of fowl adenoviruses and egg drop syndrome virus. J. Virol. Methods 1998, 73, 211–217. [Google Scholar] [CrossRef]
- Meulemans, G.; Couvreur, B.; Decaesstecker, M.; Boschmans, M.; van den Berg, T.P. Phylogenetic analysis of fowl adenoviruses. Avian Pathol. 2004, 33, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Hess, M. Aviadenovirus Infections, Diseases of Poultry; John Wiley & Sons: Hoboke, NJ, USA, 2013; pp. 290–300. [Google Scholar]
- Schachner, A.; Matos, M.; Grafl, B.; Hess, M. Fowl adenovirus-induced diseases and strategies for their control-a review on the current global situation. Avian Pathol. 2018, 47, 111–126. [Google Scholar] [CrossRef]
- Marek, A.; Kajan, G.L.; Kosiol, C.; Benko, M.; Schachner, A.; Hess, M. Genetic diversity of species Fowl aviadenovirus D and Fowl aviadenovirus E. J. Gen. Virol. 2016, 97, 2323–2332. [Google Scholar] [CrossRef]
- McFerran, J.B.; Adair, B.M. Avian adenoviruses–a review. Avian Pathol. 1977, 6, 189–217. [Google Scholar] [CrossRef]
- Zhao, J.; Zhong, Q.; Zhao, Y.; Hu, Y.X.; Zhang, G.Z. Pathogenicity and Complete Genome Characterization of Fowl Adenoviruses Isolated from Chickens Associated with Inclusion Body Hepatitis and Hydropericardium Syndrome in China. PLoS ONE 2015, 10, e0133073. [Google Scholar] [CrossRef]
- Li, L.; Luo, L.; Luo, Q.; Zhang, T.; Zhao, K.; Wang, H.; Zhang, R.; Lu, Q.; Pan, Z.; Shao, H.; et al. Genome Sequence of a Fowl Adenovirus Serotype 4 Strain Lethal to Chickens, Isolated from China. Genome Announc. 2016, 4, e00140-16. [Google Scholar] [CrossRef]
- Liu, Y.; Wan, W.; Gao, D.; Li, Y.; Yang, X.; Liu, H.; Yao, H.; Chen, L.; Wang, C.; Zhao, J. Genetic characterization of novel fowl aviadenovirus 4 isolates from outbreaks of hepatitis-hydropericardium syndrome in broiler chickens in China. Emerg. Microbes Infect. 2016, 5, e117. [Google Scholar] [CrossRef]
- Marek, A.; Kosiol, C.; Harrach, B.; Kajan, G.L.; Schlotterer, C.; Hess, M. The first whole genome sequence of a Fowl adenovirus B strain enables interspecies comparisons within the genus Aviadenovirus. Vet. Microbiol. 2013, 166, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Schachner, A.; Marek, A.; Grafl, B.; Hess, M. Detailed molecular analyses of the hexon loop-1 and fibers of fowl aviadenoviruses reveal new insights into the antigenic relationship and confirm that specific genotypes are involved in field outbreaks of inclusion body hepatitis. Vet. Microbiol. 2016, 186, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, H.; Shimizu, F.; Tsubahara, H. Avian Adenovirus: Its Properties and Serological Classification. Natl. Inst. Anim. Health Q. (Tokyo) 1964, 4, 183–193. [Google Scholar] [PubMed]
- Grafl, B.; Aigner, F.; Liebhart, D.; Marek, A.; Prokofieva, I.; Bachmeier, J.; Hess, M. Vertical transmission and clinical signs in broiler breeders and broilers experiencing adenoviral gizzard erosion. Avian Pathol. 2012, 41, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Hess, M.; Raue, R.; Prusas, C. Epidemiological studies on fowl adenoviruses isolated from cases of infectious hydropericardium. Avian Pathol. 1999, 28, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.J.; Calnek, B.W. Isolation and Classification of Avian Enteric Cytopathogenic Agents. Avian Dis. 1962, 6, 51–58. [Google Scholar] [CrossRef]
- Khanna, P.N. Occurrence of Avian Adenoviruses in Hungary. Acta Vet. Acad. Sci. Hung. 1966, 16, 351–356. [Google Scholar]
- Hess, M.; Prusas, C.; Bergmann, V.; Mazaheri, A.; Raue, R. Epizootiologische Untersuchungen über Hühneradenoviren. Berl. Münch. Tierärztl. Wochenschr. 2000, 113, 202–208. [Google Scholar]
- Yates, V.J.; Fry, D.E. Observations on a chicken embryo lethal orphan (CELO) virus. Am. J. Vet. Res. 1957, 18, 657–660. [Google Scholar]
- Matczuk, A.K.; Niczyporuk, J.S.; Kuczkowski, M.; Wozniakowski, G.; Nowak, M.; Wieliczko, A. Whole genome sequencing of Fowl aviadenovirus A-a causative agent of gizzard erosion and ulceration, in adult laying hens. Infect. Genet. Evol. 2017, 48, 47–53. [Google Scholar] [CrossRef]
- Thanasut, K.; Fujino, K.; Taharaguchi, M.; Taharaguchi, S.; Shimokawa, F.; Murakami, M.; Takase, K. Genome Sequence of Fowl Aviadenovirus A Strain JM1/1, Which Caused Gizzard Erosions in Japan. Microbiol. Resour. Ann. 2017, 5, e00749-17. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Tian, Y.M.; Han, X.X.; Yang, X.; Wang, H.N. Complete genome sequence and pathogenicity of fowl adenovirus serotype 4 involved in hydropericardium syndrome in Southwest China. Microb. Pathog. 2018, 117, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Vera-Hernandez, P.F.; Morales-Garzon, A.; Cortes-Espinosa, D.V.; Galiote-Flores, A.; Garcia-Barrera, L.J.; Rodriguez-Galindo, E.T.; Toscano-Contreras, A.; Lucio-Decanini, E.; Absalon, A.E. Clinicopathological characterization and genomic sequence differences observed in a highly virulent fowl Aviadenovirus serotype 4. Avian Pathol. 2016, 45, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Absalon, A.E.; Morales-Garzon, A.; Vera-Hernandez, P.F.; Cortes-Espinosa, D.V.; Uribe-Ochoa, S.M.; Garcia, L.J.; Lucio-Decanini, E. Complete genome sequence of a non-pathogenic strain of Fowl Adenovirus serotype 11: Minimal genomic differences between pathogenic and nonpathogenic viruses. Virology 2017, 501, 63–69. [Google Scholar] [CrossRef]
- Slaine, P.D.; Ackford, J.G.; Kropinski, A.M.; Kozak, R.A.; Krell, P.J.; Nagy, E. Molecular characterization of pathogenic and nonpathogenic fowl aviadenovirus serotype 11 isolates. Can. J. Microbiol. 2016, 62, 993–1002. [Google Scholar] [CrossRef]
- Izquierdo-Lara, R.; Calderon, K.; Chumbe, A.; Montesinos, R.; Montalvan, A.; Gonzalez, A.E.; Icochea, E.; Fernandez-Diaz, M. Complete Genome Sequence of Fowl Aviadenovirus Serotype 8b Isolated in South America. Microbiol. Resour. Ann. 2016, 4, e01174-16. [Google Scholar] [CrossRef]
- Juliana, M.; Nurulfiza, I.; Hair-Bejo, M.; Omar, A.; Aini, I. Tropical Agricultural Science. Pertanika J. Trop. Agric. Sci. 2014, 37, 483–497. [Google Scholar]
- Schat, K.; Sellers, H.A. Cell culture methods. In A Laboratory Manual for the Isolation and Identification of Avian Pathogens, 5th ed.; Dufour-Zavala, L., Swayne, D.E., Glisson, J.R., Pearson, J.E., Reed, W.M., Jackwood, M.W., Woolcock, P.R., Eds.; OmniPress: Madison, WI, USA, 2008; pp. 195–203. [Google Scholar]
- Marek, A.; Nolte, V.; Schachner, A.; Berger, E.; Schlotterer, C.; Hess, M. Two fiber genes of nearly equal lengths are a common and distinctive feature of Fowl adenovirus C members. Vet. Microbiol. 2012, 156, 411–417. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 2016, 32, 1933–1942. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [PubMed]
- Robinson, D.F.; Foulds, L.R. Comparison of Phylogenetic Trees. Math. Biosci. 1981, 53, 131–147. [Google Scholar] [CrossRef]
- Obenauer, J.C.; Denson, J.; Mehta, P.K.; Su, X.P.; Mukatira, S.; Finkelstein, D.B.; Xu, X.Q.; Wang, J.H.; Ma, J.; Fan, Y.P.; et al. Large-scale sequence analysis of avian influenza isolates. Science 2006, 311, 1576–1580. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P. Recombination detection and analysis using RDP3. Methods Mol. Biol. 2009, 537, 185–205. [Google Scholar]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retrovir. 2005, 21, 98–102. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, P.; Lett, J.M.; Varsani, A.; Martin, D.P. Widely Conserved Recombination Patterns among Single-Stranded DNA Viruses. J. Virol. 2009, 83, 2697–2707. [Google Scholar] [CrossRef] [PubMed]
- Heath, L.; van der Walt, E.; Varsani, A.; Martin, D.P. Recombination patterns in aphthoviruses mirror those found in other picornaviruses. J. Virol. 2006, 80, 11827–11832. [Google Scholar] [CrossRef] [PubMed]
- Steel, M.A.; Penny, D. Distributions of tree comparison metrics—Some new results. Syst. Biol. 1993, 42, 126–141. [Google Scholar]
- McFerran, J.B.; Clarke, J.K.; Connor, T.J. Serological classification of avian adenoviruses. Arch. Ges. Virusforsch. 1972, 39, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Norrby, E. The structural and functional diversity of Adenovirus capsid components. J. Gen. Virol. 1969, 5, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Archetti, I.; Horsfall, F.L. Persistent Antigenic Variation of Influenza a Viruses after Incomplete Neutralization in Ovo with Heterologous Immune Serum. J. Exp. Med. 1950, 92, 441–462. [Google Scholar] [CrossRef]
- Schonewille, E.; Jaspers, R.; Paul, G.; Hess, M. Specific-pathogen-free chickens vaccinated with a live FAdV-4 vaccine are fully protected against a severe challenge even in the absence of neutralizing antibodies. Avian Dis. 2010, 54, 905–910. [Google Scholar] [CrossRef]
- Sheppard, M.; McCoy, R.J.; Werner, W. Genomic mapping and sequence analysis of the fowl adenovirus serotype 10 hexon gene. J. Gen. Virol. 1995, 76, 2595–2600. [Google Scholar] [CrossRef]
- Lukashev, A.N.; Ivanova, O.E.; Eremeeva, T.P.; Iggo, R.D. Evidence of frequent recombination among human adenoviruses. J. Gen. Virol. 2008, 89, 380–388. [Google Scholar] [CrossRef]
- Robinson, C.M.; Rajaiya, J.; Walsh, M.P.; Seto, D.; Dyer, D.W.; Jones, M.S.; Chodosh, J. Computational analysis of human adenovirus type 22 provides evidence for recombination among species D human adenoviruses in the penton base gene. J. Virol. 2009, 83, 8980–8985. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Seto, D.; Jones, M.S.; Dyer, D.W.; Chodosh, J. Molecular evolution of human species D adenoviruses. Infect. Genet. Evol. 2011, 11, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Robinson, C.M.; Dehghan, S.; Schmidt, T.; Seto, D.; Jones, M.S.; Dyer, D.W.; Chodosh, J. Overreliance on the Hexon Gene, Leading to Misclassification of Human Adenoviruses. J. Virol. 2012, 86, 4693–4695. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Robinson, C.M.; Dehghan, S.; Jones, M.S.; Dyer, D.W.; Seto, D.; Chodosh, J. Homologous Recombination in E3 Genes of Human Adenovirus Species D. J. Virol. 2013, 87, 12481–12488. [Google Scholar] [CrossRef]
- Gonzalez, G.; Koyanagi, K.O.; Aoki, K.; Kitaichi, N.; Ohno, S.; Kaneko, H.; Ishida, S.; Watanabe, H. Intertypic modular exchanges of genomic segments by homologous recombination at universally conserved segments in human adenovirus species D. Gene 2014, 547, 10–17. [Google Scholar] [CrossRef]
- Gonzalez, G.; Bair, C.R.; Lamson, D.M.; Watanabe, H.; Panto, L.; Carr, M.J.; Kajon, A.E. Genomic characterization of human adenovirus type 4 strains isolated worldwide since 1953 identifies two separable phylogroups evolving at different rates from their most recent common ancestor. Virology 2019, 538, 11–23. [Google Scholar] [CrossRef]
- Nagy, M.; Nagy, E.; Tuboly, T. Sequence analysis of porcine adenovirus serotype 5 fibre gene: Evidence for recombination. Virus Genes 2002, 24, 181–185. [Google Scholar] [CrossRef]
- Wong, M.; Woolford, L.; Hasan, N.H.; Hemmatzadeh, F. A Novel Recombinant Canine Adenovirus Type 1 Detected from Acute Lethal Cases of Infectious Canine Hepatitis. Viral Immunol. 2017, 30, 258–263. [Google Scholar] [CrossRef]
- Iglesias-Caballero, M.; Juste, J.; Vazquez-Moron, S.; Falcon, A.; Aznar-Lopez, C.; Ibanez, C.; Pozo, F.; Ruiz, G.; Berciano, J.M.; Garin, I.; et al. New Adenovirus Groups in Western Palaearctic Bats. Viruses 2018, 10, 443. [Google Scholar] [CrossRef]
- Das, S.; Fearnside, K.; Sarker, S.; Forwood, J.K.; Raidal, S.R. A novel pathogenic aviadenovirus from red-bellied parrots (Poicephalus rufiventris) unveils deep recombination events among avian host lineages. Virology 2017, 502, 188–197. [Google Scholar] [CrossRef]
- Chiocca, S.; Kurzbauer, R.; Schaffner, G.; Baker, A.; Mautner, V.; Cotten, M. The complete DNA sequence and genomic organization of the avian adenovirus CELO. J. Virol. 1996, 70, 2939–2949. [Google Scholar] [PubMed]
- Grgic, H.; Yang, D.H.; Nagy, E. Pathogenicity and complete genome sequence of a fowl adenovirus serotype 8 isolate. Virus Res. 2011, 156, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Benko, M.; Harrach, B. Genetic content and evolution of adenoviruses. J. Gen. Virol. 2003, 84, 2895–2908. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.D.; Nagy, E. Coding potential and transcript analysis of fowl adenovirus 4: Insight into upstream ORFs as common sequence features in adenoviral transcripts. J. Gen. Virol. 2011, 92, 1260–1272. [Google Scholar] [CrossRef] [PubMed]
- Darr, S.; Madisch, I.; Hofmayer, S.; Rehren, F.; Heim, A. Phylogeny and primary structure analysis of fiber shafts of all human adenovirus types for rational design of adenoviral gene-therapy vectors. J. Gen. Virol. 2009, 90, 2849–2854. [Google Scholar] [CrossRef] [PubMed]
- Kajan, G.L.; Affranio, I.; Tothne Bistyak, A.; Kecskemeti, S.; Benko, M. An emerging new fowl adenovirus genotype. Heliyon 2019, 5, e01732. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.; Grafl, B.; Liebhart, D.; Schwendenwein, I.; Hess, M. Selected clinical chemistry analytes correlate with the pathogenesis of inclusion body hepatitis experimentally induced by fowl aviadenoviruses. Avian Pathol. 2016, 45, 520–529. [Google Scholar] [CrossRef]
- Calnek, B.W.; Cowen, B.S. Adenoviruses of Chickens-Serologic Groups. Avian Dis. 1975, 19, 91–103. [Google Scholar] [CrossRef]
- McFerran, J.; Connor, T. Further studies on the classification of fowl adenoviruses. Avian Dis. 1977, 21, 585–595. [Google Scholar] [CrossRef]
- Steer, P.A.; O’Rourke, D.; Ghorashi, S.A.; Noormohammadi, A.H. Application of high-resolution melting curve analysis for typing of fowl adenoviruses in field cases of inclusion body hepatitis. Aust. Vet. J. 2011, 89, 184–192. [Google Scholar] [CrossRef]
- Hess, M.; Prusas, C.; Vereecken, M.; De Herdt, P. Isolation of fowl adenoviruses serotype 4 from pigeons with hepatic necrosis. Berl. Münch. Tierärztl. Wochenschr. 1998, 111, 140–142. [Google Scholar] [PubMed]
- Gonzalez, G.; Koyanagi, K.O.; Aoki, K.; Watanabe, H. Interregional Coevolution Analysis Revealing Functional and Structural Interrelatedness between Different Genomic Regions in Human Mastadenovirus D. J. Virol. 2015, 89, 6209–6217. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benko, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schachner, A.; Gonzalez, G.; Endler, L.; Ito, K.; Hess, M. Fowl Adenovirus (FAdV) Recombination with Intertypic Crossovers in Genomes of FAdV-D and FAdV-E, Displaying Hybrid Serological Phenotypes. Viruses 2019, 11, 1094. https://doi.org/10.3390/v11121094
Schachner A, Gonzalez G, Endler L, Ito K, Hess M. Fowl Adenovirus (FAdV) Recombination with Intertypic Crossovers in Genomes of FAdV-D and FAdV-E, Displaying Hybrid Serological Phenotypes. Viruses. 2019; 11(12):1094. https://doi.org/10.3390/v11121094
Chicago/Turabian StyleSchachner, Anna, Gabriel Gonzalez, Lukas Endler, Kimihito Ito, and Michael Hess. 2019. "Fowl Adenovirus (FAdV) Recombination with Intertypic Crossovers in Genomes of FAdV-D and FAdV-E, Displaying Hybrid Serological Phenotypes" Viruses 11, no. 12: 1094. https://doi.org/10.3390/v11121094
APA StyleSchachner, A., Gonzalez, G., Endler, L., Ito, K., & Hess, M. (2019). Fowl Adenovirus (FAdV) Recombination with Intertypic Crossovers in Genomes of FAdV-D and FAdV-E, Displaying Hybrid Serological Phenotypes. Viruses, 11(12), 1094. https://doi.org/10.3390/v11121094