Seasonal Dynamics of Algae-Infecting Viruses and Their Inferred Interactions with Protists

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. High-Throughput Sequencing
2.3. Bioinformatic Pipeline
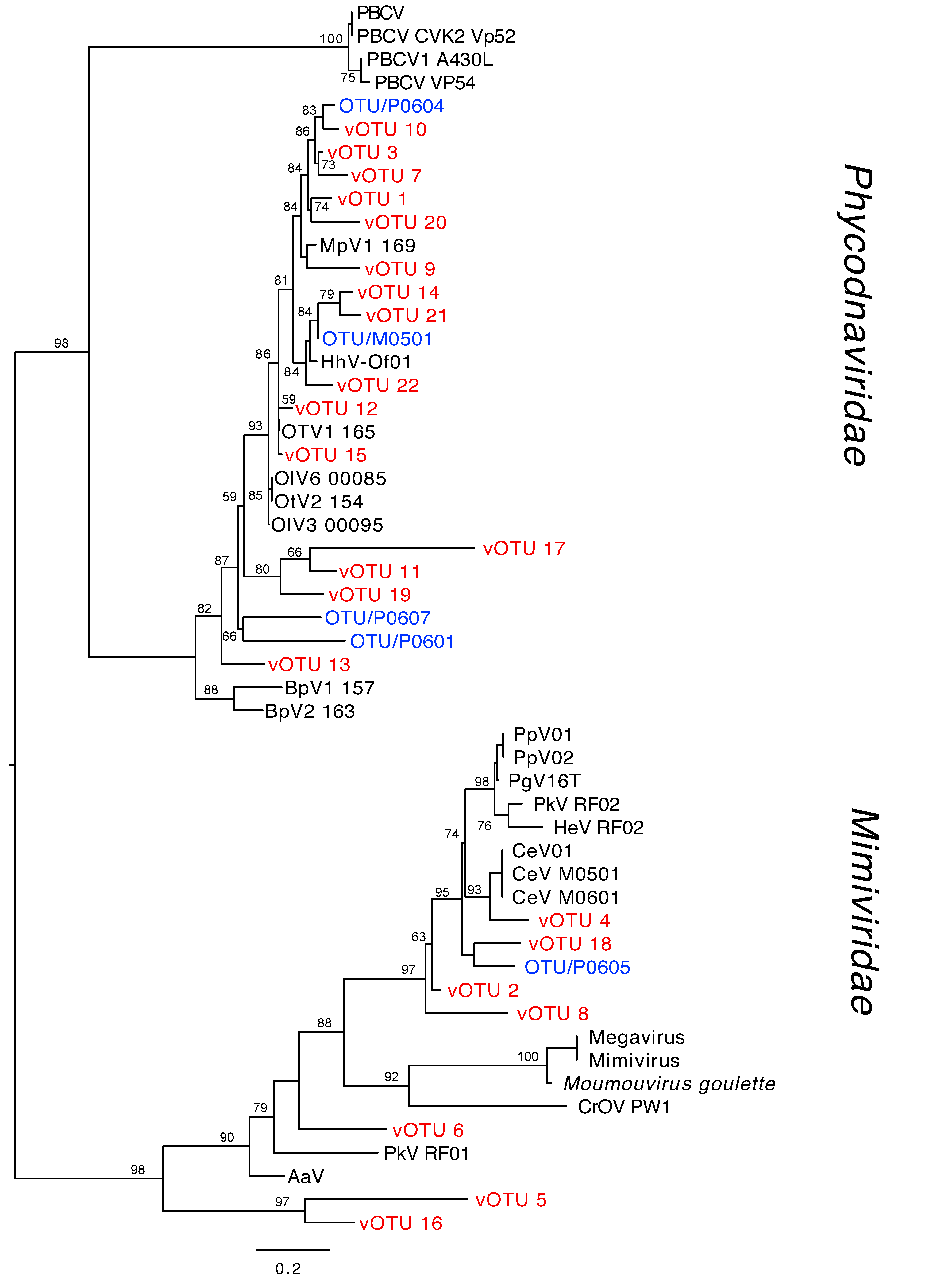
2.4. Taxonomic Classification and Phylogenetic Analyses
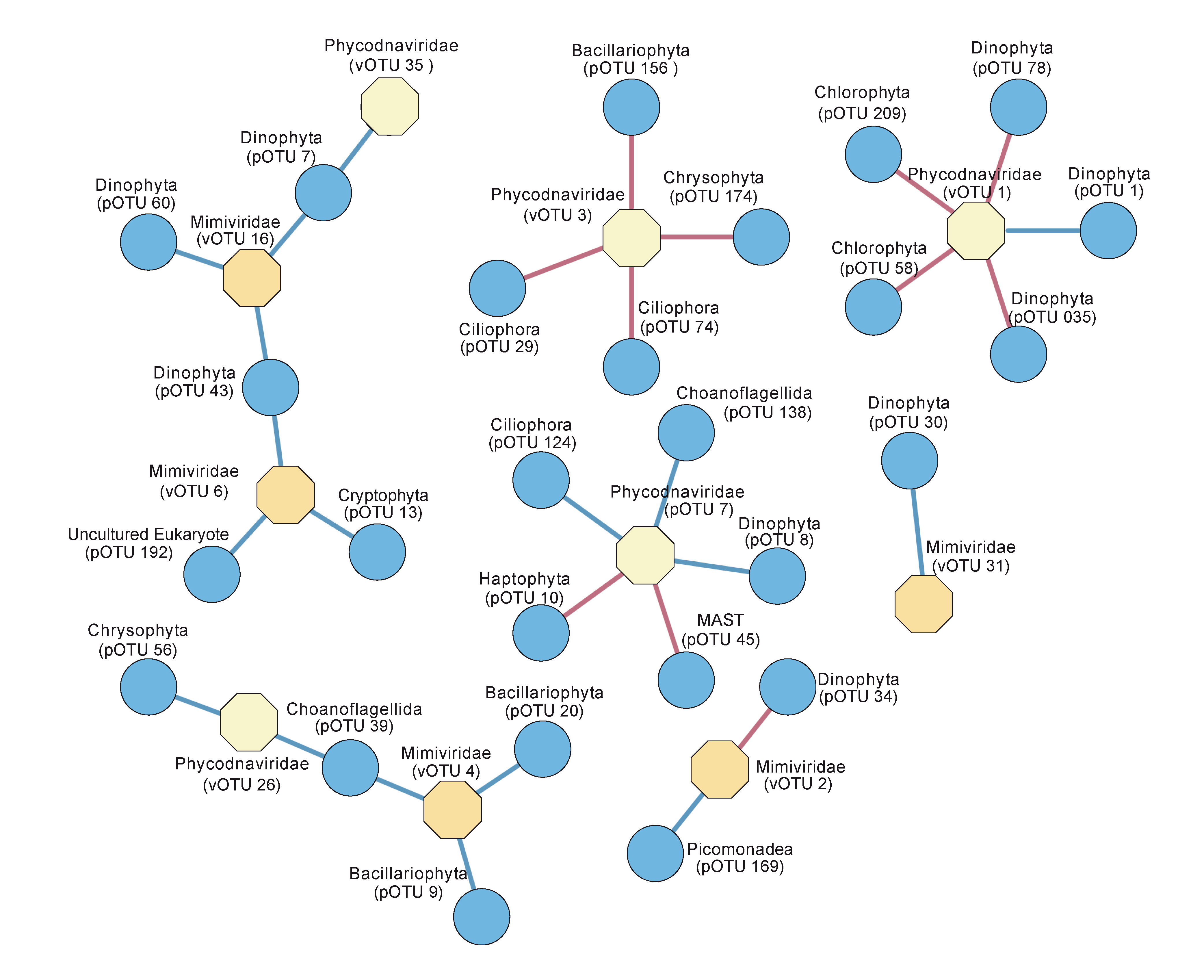
2.5. Network Construction
3. Results
3.1. Virus Diversity
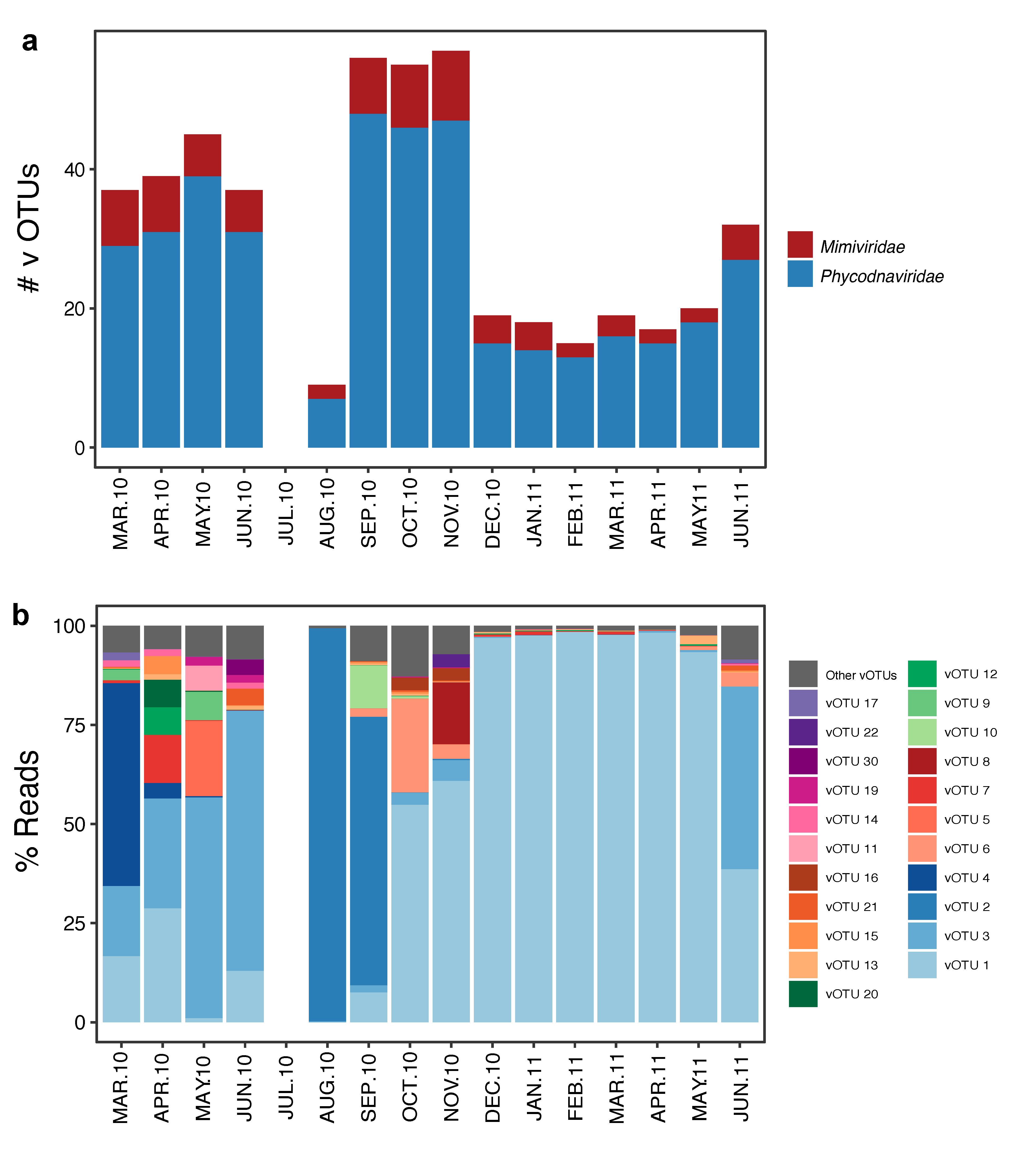
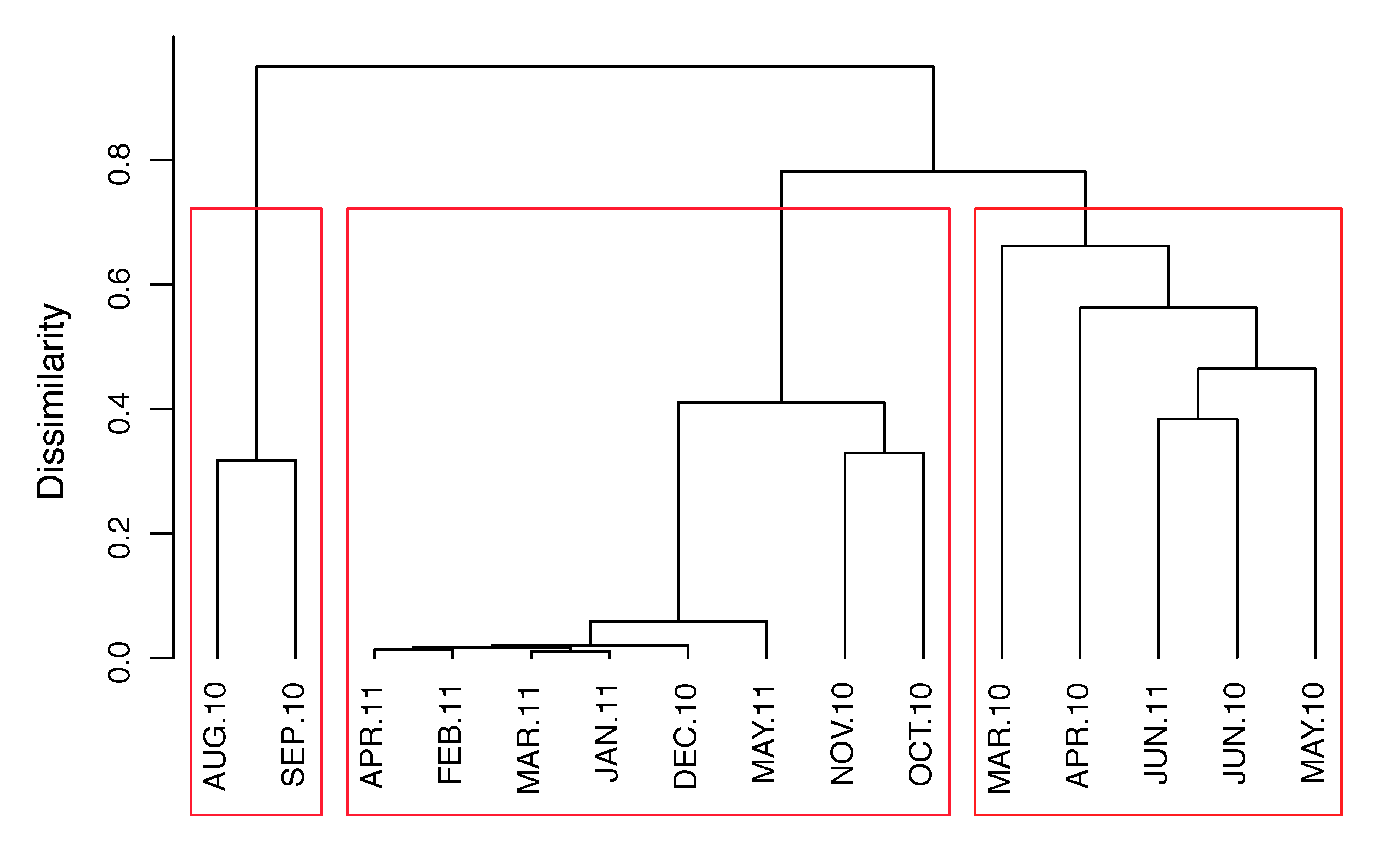
3.2. Temporal Variation
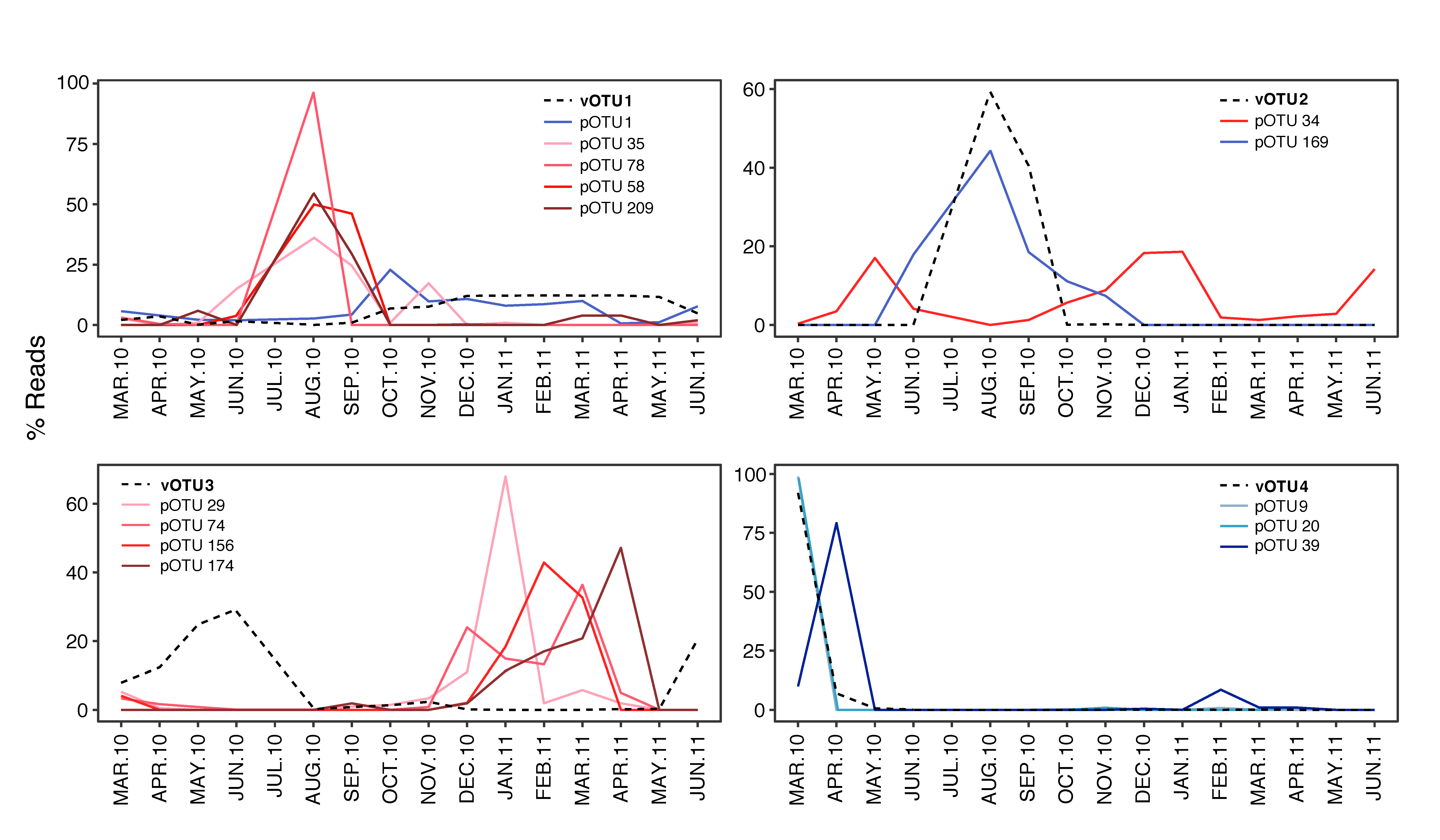
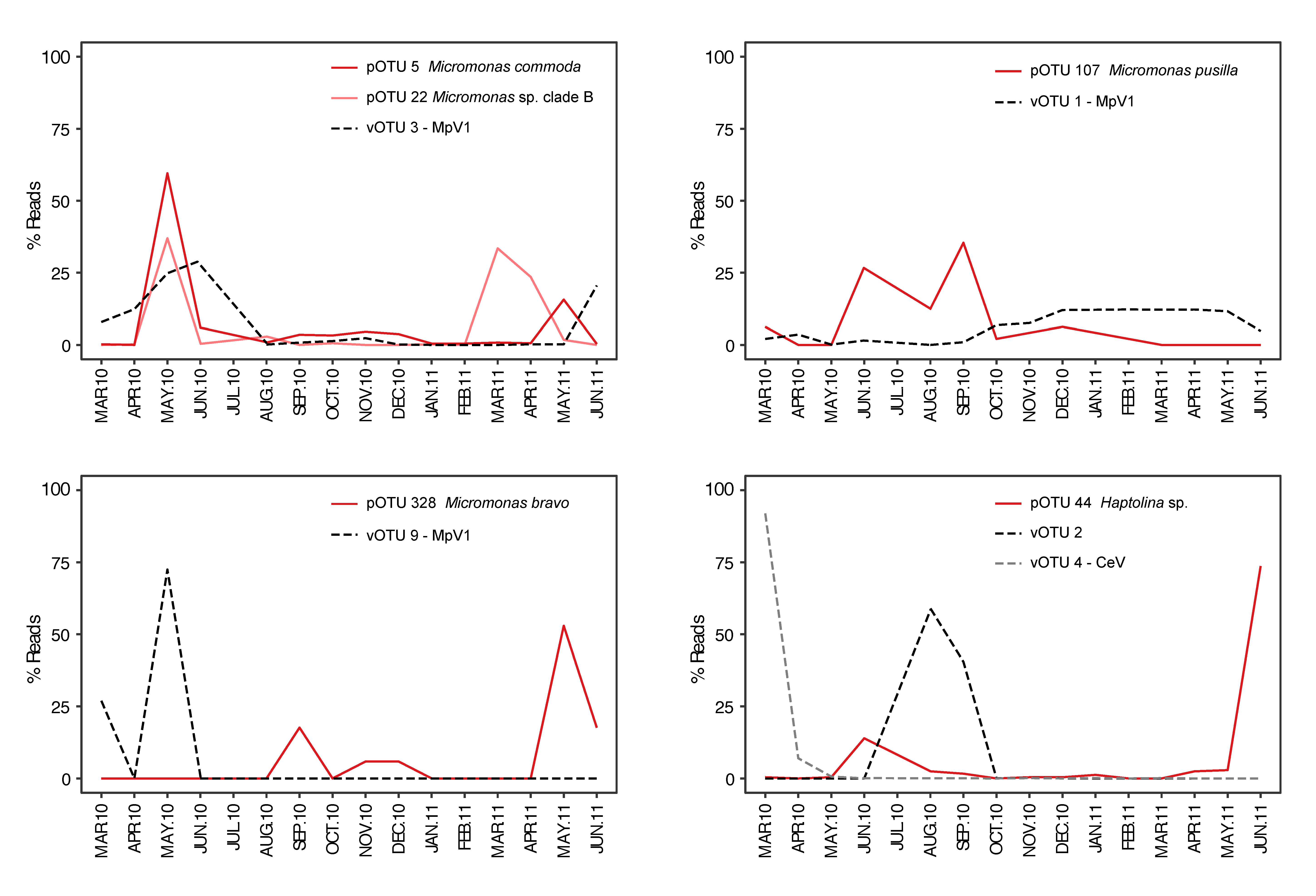
3.3. Temporal Variation of Viruses and their Potential Hosts
4. Discussion
4.1. Viral diversity
4.2. Succession of Viral OTUs and Potential Hosts
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bergh, Ø.; Børsheim, K.Y.; Bratbak, G.; Heldal, M. High abundance of viruses found in aquatic environments. Nature 1989, 340, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Boxshall, G.A. La vida en suspensión: El plancton. In La Exploración de la Biodiversidad Marina: Desafíos Científicos y Tecnológicos; Duarte, C.M., Ed.; Fundación BBVA: Bilbao, Spain, 2006; pp. 93–115. ISBN 84-96515-26-5. [Google Scholar]
- Thingstad, T.; Lignell, R. Theoretical models for the control of bacterial growth rate, abundance, diversity and carbon demand. Aquat. Microb. Ecol. 1997, 13, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Thingstad, T.F. Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol. Oceanogr. 2000, 45, 1320–1328. [Google Scholar] [CrossRef]
- Bratbak, G.; Thingstad, F.; Heldal, M. Viruses and the microbial loop. Microb. Ecol. 1994, 28, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.W.; Suttle, C.A. Viruses and nutrient cycles in the sea. BioScience 1999, 49, 781–788. [Google Scholar] [CrossRef]
- Guidi, L.; Bittner, L.; Eveillard, D.; Larhlimi, A.; Roux, S.; Darzi, Y.; Audic, S.; Berline, L.; Brum, J.R.; Coelho, L.P.; et al. Plankton networks driving carbon export in the oligotrophic ocean. Nature 2016, 532, 465–470. [Google Scholar] [CrossRef] [Green Version]
- Nissimov, J.I.; Vandzura, R.; Johns, C.T.; Natale, F.; Haramaty, L.; Bidle, K.D. Dynamics of transparent exopolymer particle production and aggregation during viral infection of the coccolithophore, Emiliania huxleyi: Dynamics of TEP production and aggregation. Environ. Microbiol. 2018, 20, 2880–2897. [Google Scholar] [CrossRef]
- Laber, C.P.; Hunter, J.E.; Carvalho, F.; Collins, J.R.; Hunter, E.J.; Schieler, B.M.; Boss, E.; More, K.; Frada, M.; Thamatrakoln, K.; et al. Coccolithovirus facilitation of carbon export in the North Atlantic. Nat. Microbiol. 2018, 3, 537–547. [Google Scholar] [CrossRef]
- Yamada, Y.; Tomaru, Y.; Fukuda, H.; Nagata, T. Aggregate formation during the viral lysis of a marine diatom. Front. Mar. Sci. 2018, 5, 167. [Google Scholar] [CrossRef]
- Dunigan, D.D.; Fitzgerald, L.A.; Van Etten, J.L. Phycodnaviruses: A peek at genetic diversity. Virus Res. 2006, 117, 119–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brussaard, C.P.D.; Martinez-Martinez, J. Algal bloom viruses. Plant Viruses 2008, 1, 1–13. [Google Scholar]
- Claverie, J.M.; Abergel, C. Mimiviridae: An expanding family of highly diverse large dsDNA viruses infecting a wide phylogenetic range of aquatic eukaryotes. Viruses 2018, 10, 506. [Google Scholar] [CrossRef]
- Boyer, M.; Madoui, M.A.; Gimenez, G.; La Scola, B.; Raoult, D. Phylogenetic and phyletic studies of informational genes in genomes highlight existence of a 4th domain of life including giant viruses. PLoS ONE 2010, 5, e15530. [Google Scholar] [CrossRef]
- Iyer, L.M.; Aravind, L.; Koonin, E.V. Common origin of four diverse families of large eukaryotic DNA viruses. J. Virol. 2001, 75, 11720–11734. [Google Scholar] [CrossRef]
- Gallot-Lavallée, L.; Pagarete, A.; Legendre, M.; Santini, S.; Sandaa, R.A.; Himmelbauer, H.; Ogata, H.; Bratbak, G.; Claverie, J.M. The 474-kilobase-pair complete genome sequence of CeV-01B, a virus infecting Haptolina (Chrysochromulina) ericina (Prymnesiophyceae). Genome Announc. 2015, 3, e01413–e01415. [Google Scholar] [CrossRef]
- Johannessen, T.V.; Bratbak, G.; Larsen, A.; Ogata, H.; Egge, E.S.; Edvardsen, B.; Eikrem, W.; Sandaa, R.A. Characterisation of three novel giant viruses reveals huge diversity among viruses infecting Prymnesiales (Haptophyta). Virology 2015, 476, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.G.; Allen, M.J.; Wilson, W.H.; Suttle, C.A. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc. Natl. Acad. Sci. USA 2010, 107, 19508–19513. [Google Scholar] [CrossRef] [Green Version]
- Mihara, T.; Koyano, H.; Hingamp, P.; Grimsley, N.; Goto, S.; Ogata, H. Taxon richness of “Megaviridae” exceeds those of bacteria and archaea in the ocean. Microbes Environ. 2018, 33, 162–171. [Google Scholar] [CrossRef]
- Brussaard, C.P.D. Viral control of phytoplankton populations—A review. J. Eukaryot. Microbiol. 2004, 51, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Baudoux, A.C. The Role of Viruses in Marine Phytoplankton Mortality. Ph.D. Thesis, Royal Netherlands Institute for Sea Research (NIOZ), Texel, The Netherland, 2007. [Google Scholar]
- Sandaa, R.A. Burden or benefit? Virus–host interactions in the marine environment. Res. Microbiol. 2008, 159, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Clerissi, C.; Grimsley, N.; Ogata, H.; Hingamp, P.; Poulain, J.; Desdevises, Y. Unveiling of the diversity of prasinoviruses (Phycodnaviridae) in marine samples by using high-throughput sequencing analyses of PCR-amplified DNA polymerase and major capsid protein genes. Appl. Environ. Microbiol. 2014, 80, 3150–3160. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.; Larsen, A.; Bratbak, G.; Pagarete, A.; Edvardsen, B.; Egge, E.; Sandaa, R.A. Seasonal dynamics of haptophytes and dsDNA algal viruses suggest complex virus-host relationship. Viruses 2017, 9, 84. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Wurch, L.L.; Alexander, H.; Dyhrman, S.T.; Gobler, C.J.; Wilhelm, S.W. Virus-host relationships of marine single-celled eukaryotes resolved from metatranscriptomics. Nat. Commun. 2017, 8, 16054. [Google Scholar] [CrossRef] [PubMed]
- Sandaa, R.A.E.; Storesund, J.; Olesin, E.; Lund Paulsen, M.; Larsen, A.; Bratbak, G.; Ray, J. Seasonality drives microbial community structure, shaping both eukaryotic and prokaryotic host–viral relationships in an arctic marine ecosystem. Viruses 2018, 10, 715. [Google Scholar] [CrossRef]
- Sandaa, R.A.; Bratbak, G. Is the virus important? And some other questions. Viruses 2018, 10, 442. [Google Scholar] [CrossRef]
- Suttle, C.A.; Chan, A.M. Dynamics and distribution of cyanophages and their effect on marine Synechococcus spp. Appl. Environ. Microbiol. 1994, 60, 3167–3174. [Google Scholar]
- Tarutani, K.; Nagasaki, K.; Itakura, S.; Yamaguchi, M. Isolation of a virus infecting the novel shellfish-killing dinoflagellate Heterocapsa circularisquama. Aquat. Microb. Ecol. 2001, 23, 103–111. [Google Scholar] [CrossRef]
- Thyrhaug, R.; Larsen, A.; Thingstad, T.F.; Bratbak, G. Stable coexistence in marine algal host-virus systems. Mar. Ecol. Prog. Ser. 2003, 254, 27–35. [Google Scholar] [CrossRef]
- Dimmock, N.J.; Easton, A.J.; Leppard, K.N. Introduction to Modern Virology, 7th ed.; John Wiley & Sons: West Sussex, UK, 2016; ISBN 978-1-119-97810-7. [Google Scholar]
- Gran-Stadniczeñko, S.; Dunthorn Egge, E.; Hostyeva, V.; Logares, R.; Eikrem, W.; Edvardsen, B. Protist diversity and seasonal dynamics in Skagerrak plankton communities as revealed by metabarcoding and microscopy. J. Eukaryot. Microbiol. 2019, 66, 494–513. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.B.; Larsen, A.; Bratbak, G.; Sandaa, R.A. Phylogenetic analysis of members of the Phycodnaviridae virus family, using amplified fragments of the major capsid protein gene. Appl. Environ. Microbiol. 2008, 74, 3048–3057. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quince, C.; Lanzen, A.; Davenport, R.J.; Turnbaugh, P.J. Removing noise from pyrosequenced amplicons. BMC Bioinform. 2011, 12, 38. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package, R package version 1.17-2; R Foundation for Statistical Computing: Vienna, Austria, 2011. [Google Scholar]
- Deng, W.; Nickle, D.C.; Learn, G.H.; Maust, B.; Mullins, J.I. ViroBLAST: A stand-alone BLAST web server for flexible queries of multiple databases and user’s datasets. Bioinformatics 2007, 23, 2334–2336. [Google Scholar] [CrossRef]
- Besemer, J.; Borodovsky, M. Heuristic approach to deriving models for gene finding. Nucleic Acids Res. 1999, 27, 3911–3920. [Google Scholar] [CrossRef] [Green Version]
- Besemer, J.; Borodovsky, M. GeneMark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005, 33, W451–W454. [Google Scholar] [CrossRef]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evolut. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 2016, 32, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Frith, M.C. Adding unaligned sequences into an existing alignment using MAFFT and LAST. Bioinformatics 2012, 28, 3144–3146. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—approximately maximum-likelihood Ttees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J. FigTree v1.4.3. 2016. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 9 November 2019).
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef]
- Kurtz, Z.D.; Müller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and compositionally robust inference of microbial ecological networks. PLOS Comput. Biol. 2015, 11, e1004226. [Google Scholar] [CrossRef]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinform. 2014, 47, 8–13. [Google Scholar] [CrossRef]
- Clerissi, C.; Desdevises, Y.; Romac, S.; Audic, S.; de Vargas, C.; Acinas, S.G.; Casotti, R.; Poulain, J.; Wincker, P.; Hingamp, P.; et al. Deep sequencing of amplified Prasinovirus and host green algal genes from an Indian Ocean transect reveals interacting trophic dependencies and new genotypes: Marine algae and their viruses in the Indian Ocean. Environ. Microbiol. Rep. 2015, 7, 979–989. [Google Scholar] [CrossRef]
- Rozon, R.M.; Short, S.M. Complex seasonality observed amongst diverse phytoplankton viruses in the Bay of Quinte, an embayment of Lake Ontario. Freshw. Biol. 2013, 58, 2648–2663. [Google Scholar] [CrossRef]
- Zhong, X.; Jacquet, S. Contrasting diversity of phycodnavirus signature genes in two large and deep western European lakes. Environ. Microbiol. 2014, 16, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Throndsen, J.; Hasle, G.R.; Tangen, K. Phytoplankton of Norwegian Coastal Waters; Almater Forlag AS: Oslo, Norway, 2007. [Google Scholar]
- Simon, N.; Foulon, E.; Grulois, D.; Six, C.; Desdevises, Y.; Latimier, M.; Le Gall, F.; Tragin, M.; Houdan, A.; Derelle, E.; et al. Revision of the genus Micromonas Manton et Parke (Chlorophyta, Mamiellophyceae), of the type species M. pusilla (Butcher) Manton & Parke and of the species M. commoda van Baren, Bachy and Worden and description of two new species based on the genetic and phenotypic characterization of cultured isolates. Protist 2017, 168, 612–635. [Google Scholar] [PubMed]
- Tragin, M.; Vaulot, D. Novel diversity within marine Mamiellophyceae (Chlorophyta) unveiled by metabarcoding. Sci. Rep. 2018, 9, 5190. [Google Scholar] [CrossRef]
- Weynberg, K.D.; Allen, M.J.; Ashelford, K.; Scanlan, D.J.; Wilson, W.H. From small hosts come big viruses: The complete genome of a second Ostreococcus tauri virus, OtV-1. Environ. Microbiol. 2009, 11, 2821–2839. [Google Scholar] [CrossRef]
- Pagarete, A.; Chow, C.E.T.; Johannessen, T.; Fuhrman, J.A.; Thingstad, T.F.; Sandaa, R.A. Strong seasonality and interannual recurrence in marine myovirus communities. Appl. Environ. Microbiol. 2013, 20, 6253–6259. [Google Scholar] [CrossRef]
- Needham, D.M.; Chow, C.E.T.; Cram, J.A.; Sachdeva, R.; Parada, A.; Fuhrman, J.A. Short-term observations of marine bacterial and viral communities: Patterns, connections and resilience. ISME J. 2013, 7, 1274–1285. [Google Scholar] [CrossRef]
- Short, S.M. The ecology of viruses that infect eukaryotic algae. Environ. Microbiol. 2012, 14, 2253–2271. [Google Scholar] [CrossRef]
- Sahlsten, E. Seasonal abundance in Skagerrak-Kattegat coastal waters and host specificity of viruses infecting the marine photosynthetic flagellate Micromonas pusilla. Aquat. Microb. Ecol. 1998, 16, 103–108. [Google Scholar] [CrossRef]
- Zingone, A.; Sarno, D.; Forlani, G. Seasonal dynamics in the abundance of Micromonas pusilla (Prasinophyceae) and its viruses in the Gulf of Naples (Mediterranean Sea). J. Plankton Res. 1999, 21, 2143–2159. [Google Scholar] [CrossRef]
- Zingone, A.; Natale, F.; Biffali, E.; Borra, M.; Forlani, G.; Sarno, D. Diversity in morphology, infectivity, molecular characteristics and induced host resistance between two viruses infecting Micromonas pusilla. Aquat. Microb. Ecol. 2006, 45, 1–14. [Google Scholar] [CrossRef]
- Goic, B.; Saleh, M.C. Living with the enemy: Viral persistent infections from a friendly viewpoint. Curr. Opin. Microbiol. 2012, 15, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Longdon, B.; Brockhurst, M.A.; Russell, C.A.; Welch, J.J.; Jiggins, F.M. The evolution and genetics of virus host shifts. PLoS Pathog. 2014, 10, e1004395. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VIRUS | PROTIST | ||||||
|---|---|---|---|---|---|---|---|
| vOTU ID | Family | Lowest Taxonomic Level | Co-Occurrence1 | pOTU ID | Supergroup | Group | Lowest Taxonomic Level |
| vOTU 1 | Phycodnaviridae | Micromonas pusilla virus (MpV1) | + | pOTU 1 | Alveolata | Dinophyta | Karenia papillonaceae |
| - | pOTU 35 | Alveolata | Dinophyta | MALV-III | |||
| - | pOTU 78 | Alveolata | Dinophyta | Uncultured dinoflagellate | |||
| - | pOTU 58 | Archaeplastida | Chlorophyta | Pycnococcus provasolii | |||
| - | pOTU 209 | Archaeplastida | Chlorophyta | Pyramimonadales | |||
| vOTU 2 | Mimiviridae | Uncultured | - | pOTU 34 | Alveolata | Dinophyta | Uncultured dinoflagellate |
| + | pOTU 169 | Picozoa | Picomonadea | Picobiliphyta | |||
| vOTU 3 | Phycodnaviridae | Micromonas pusilla virus (MpV1) | - | pOTU 74 | Alveolata | Ciliophora | Cyclotrichia |
| - | pOTU 29 | Alveolata | Ciliophora | Strombidiidae | |||
| - | pOTU 156 | Stramenopila | Bacillariophyta | Coscinodiscophyceae | |||
| - | pOTU 174 | Stramenopila | Chrysophyta | Chrysophyceae-Clade-C | |||
| vOTU 4 | Mimiviridae | Chrysochromulina ericina virus (CeV) | + | pOTU 39 | Opistokonta | Choanoflagellida | Stephanoecidae-Group-D |
| + | pOTU 20 | Stramenopila | Bacillariophyta | Pseudo-nitzschia multiseries | |||
| + | pOTU 9 | Stramenopila | Bacillariophyta | Thalassiosira nordenskioeldii | |||
| vOTU 6 | Mimiviridae | Uncultured | + | pOTU 13 | Hacrobia | Cryptophyta | Teleaulax gracilis |
| + | pOTU 192 | Uncultured eukaryote | Uncultured Eukaryote | Uncultured eukaryote | |||
| vOTU 7 | Phycodnaviridae | Micromonas pusilla virus (MpV1) | + | pOTU 124 | Alveolata | Ciliophora | Haptoria |
| + | pOTU 8 | Alveolata | Dinophyta | Gyrodinium helveticum | |||
| - | pOTU 10 | Hacrobia | Haptophyta | Emiliania huxleyi | |||
| + | pOTU 138 | Opistokonta | Choanoflagellida | Stephanoecidae-Group-D | |||
| - | pOTU 45 | Stramenopila | MAST | MAST-1C | |||
| vOTU 16 | Mimiviridae | Uncultured | + | pOTU 7 | Alveolata | Dinophyta | Lepidodinium chlorophorum/L. viride |
| + | pOTU 60 | Alveolata | Dinophyta | MALV-III | |||
| + | pOTU 43 | Alveolata | Dinophyta | Uncultured dinoflagellate | |||
| vOTU 26 | Phycodnaviridae | Uncultured | + | pOTU 39 | Opistokonta | Choanoflagellida | Stephanoecidae-Group-D |
| + | pOTU 56 | Stramenopila | Chrysophyta | Chrysophyceae-Clade-C | |||
| vOTU 31 | Mimiviridae | Chrysochromulina ericina virus (CeV) | + | pOTU 30 | Alveolata | Dinophyta | Uncultured dinoflagellate |
| vOTU 35 | Phycodnaviridae | Micromonas pusilla virus (MpV1) | + | pOTU 7 | Alveolata | Dinophyta | Lepidodinium chlorophorum/L. viride |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gran-Stadniczeñko, S.; Krabberød, A.K.; Sandaa, R.-A.; Yau, S.; Egge, E.; Edvardsen, B. Seasonal Dynamics of Algae-Infecting Viruses and Their Inferred Interactions with Protists. Viruses 2019, 11, 1043. https://doi.org/10.3390/v11111043
Gran-Stadniczeñko S, Krabberød AK, Sandaa R-A, Yau S, Egge E, Edvardsen B. Seasonal Dynamics of Algae-Infecting Viruses and Their Inferred Interactions with Protists. Viruses. 2019; 11(11):1043. https://doi.org/10.3390/v11111043
Chicago/Turabian StyleGran-Stadniczeñko, Sandra, Anders K. Krabberød, Ruth-Anne Sandaa, Sheree Yau, Elianne Egge, and Bente Edvardsen. 2019. "Seasonal Dynamics of Algae-Infecting Viruses and Their Inferred Interactions with Protists" Viruses 11, no. 11: 1043. https://doi.org/10.3390/v11111043