Zika Virus Non-Structural Protein NS5 Inhibits the RIG-I Pathway and Interferon Lambda 1 Promoter Activation by Targeting IKK Epsilon

 and
and 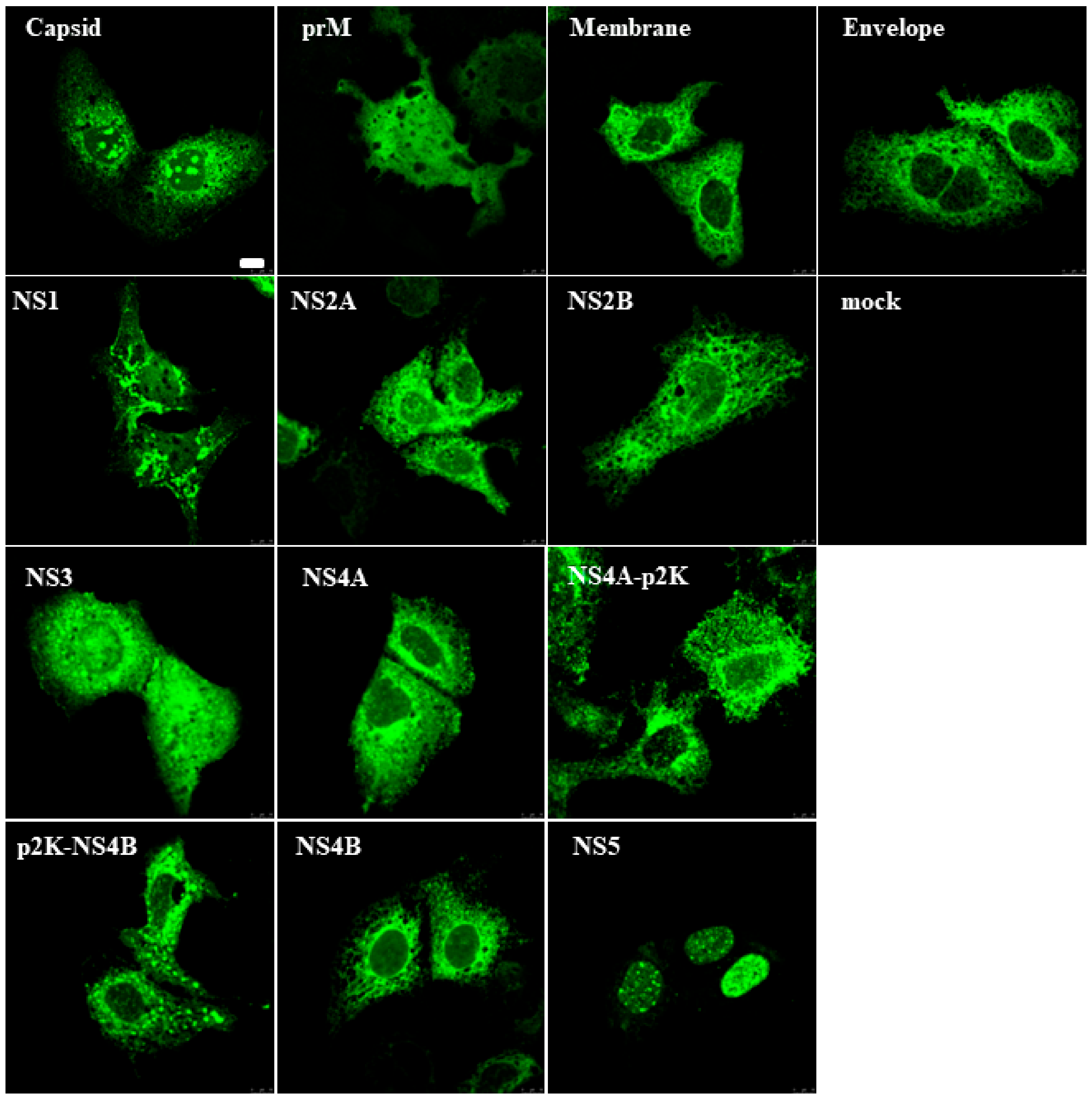{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cloning of ZIKV Genes and Construction of Expression Plasmids
2.2. Antibodies
2.3. Cells
2.4. Transfections, Cell Stimulations, and Infections
2.5. Reporter Gene Assay
2.6. Immunoblotting
2.7. Immunofluorescence Microscopy
2.8. Production of GST Fusion Proteins in Sf9 Cells and GST-Pull-Down Assay
2.9. Statistical Analyses
3. Results
3.1. Zika Virus Protein Expression in Mammalian Cells
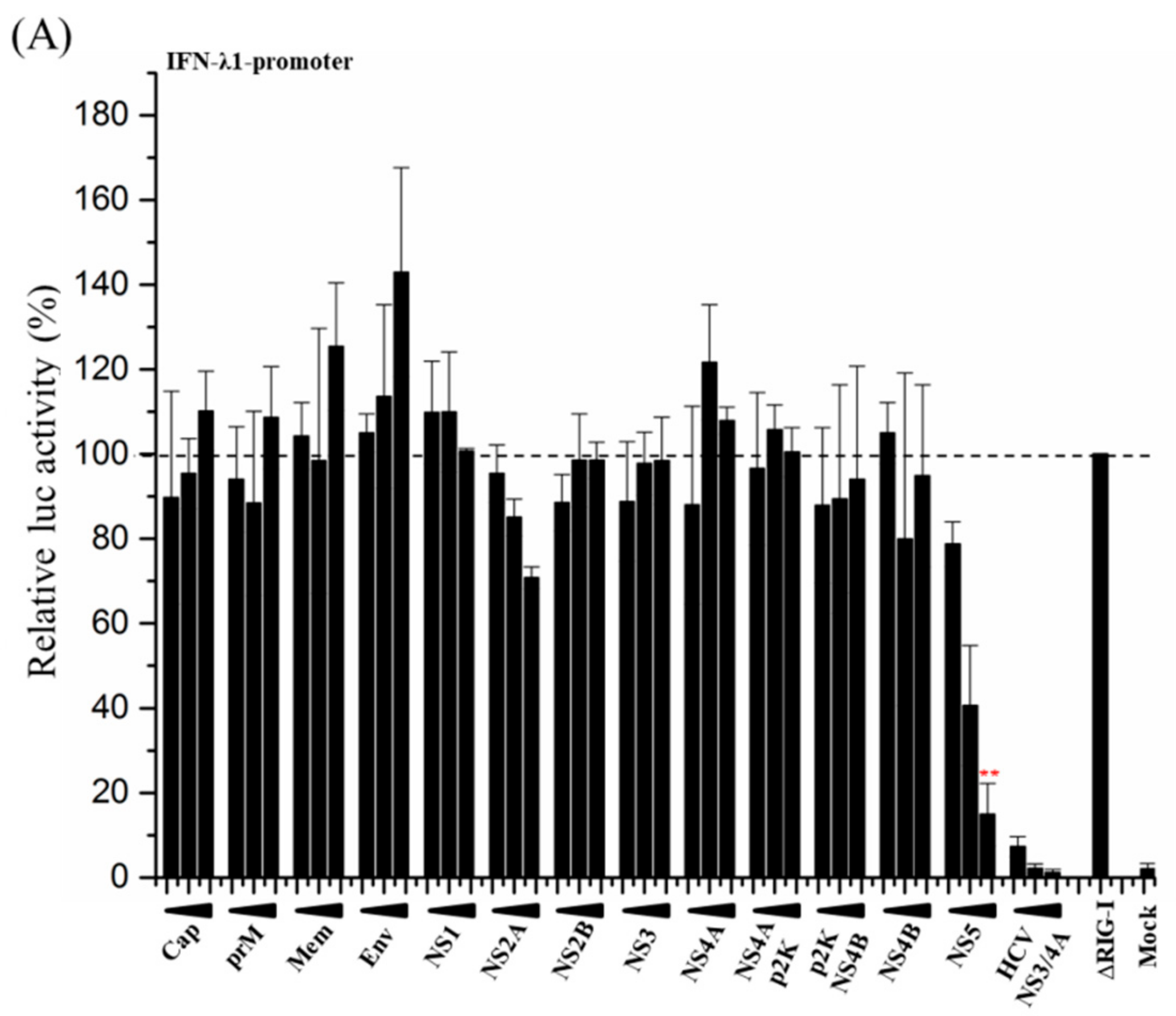
3.2. ZIKV NS5 Strongly Inhibits the Activation of IFN-λ1 and IFN-β Promoters
3.3. ZIKV NS5 of Isolate FB-GWUH-2016 Inhibits MxA-Promoter Activation
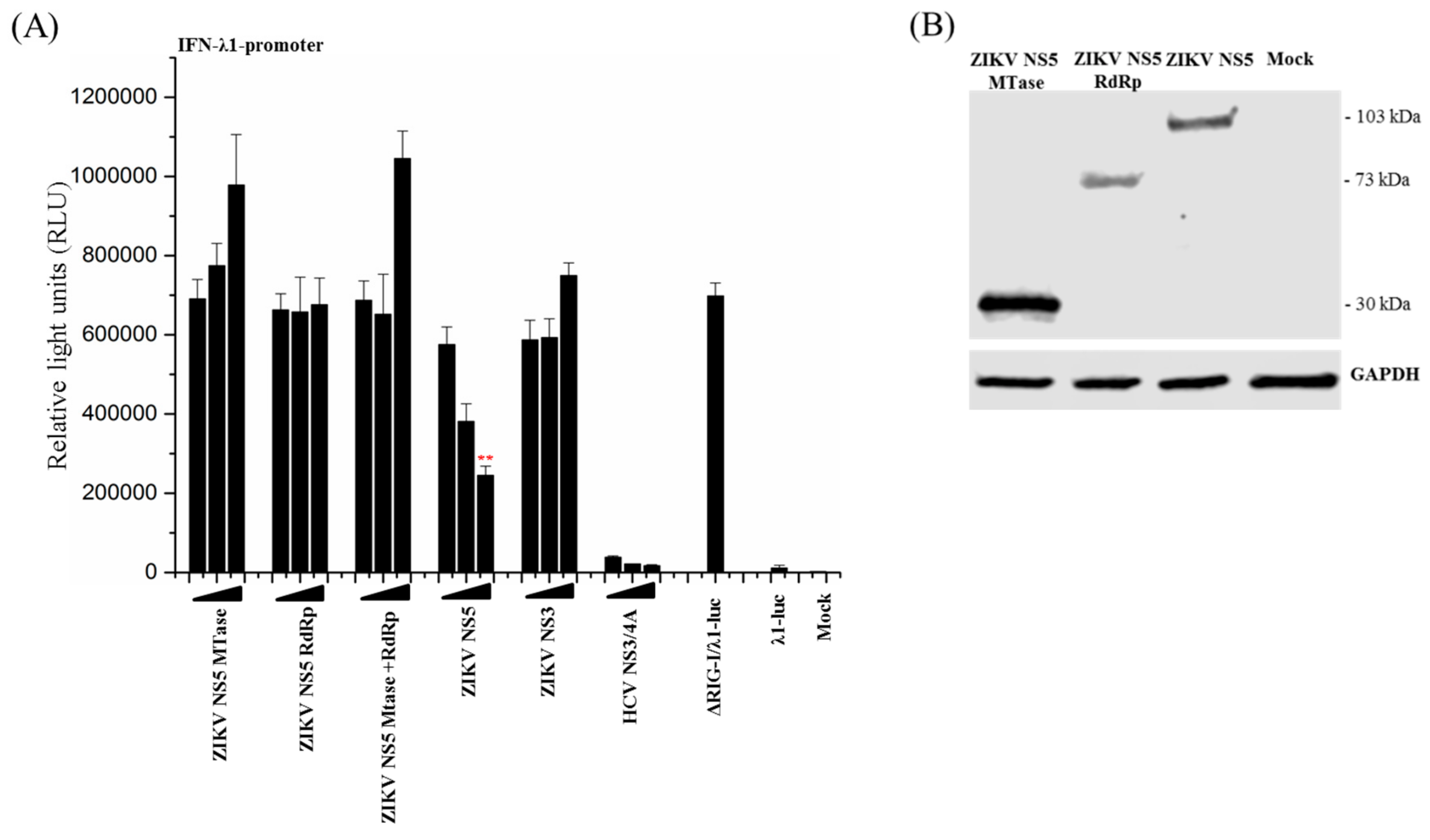
3.4. Full-Length ZIKV NS5 is Required for Its Inhibitory Activity on the RIG-I Pathway
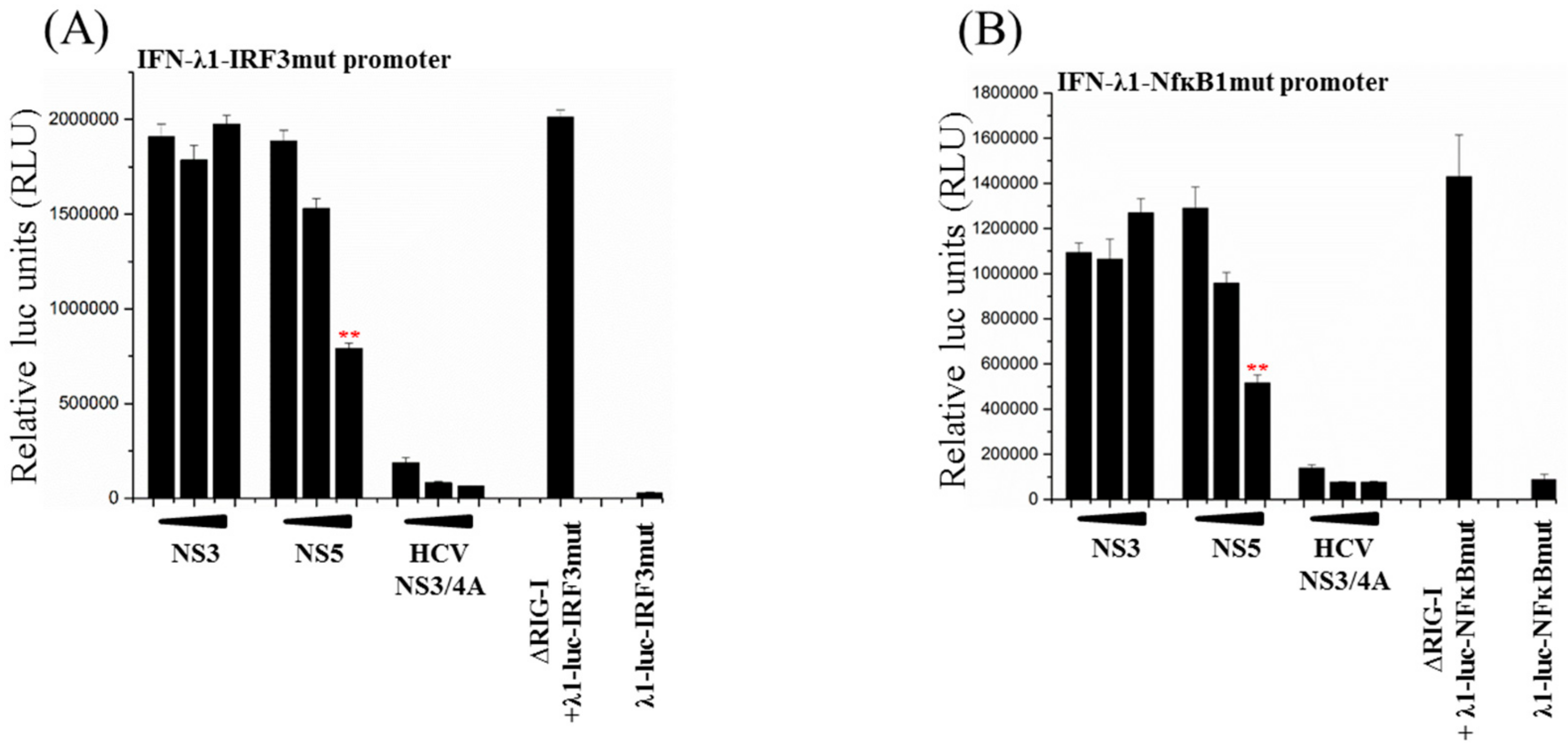
3.5. Activation of Both IRF3 and NF-ĸB Transcription Factors Are Inhibited by ZIKV NS5
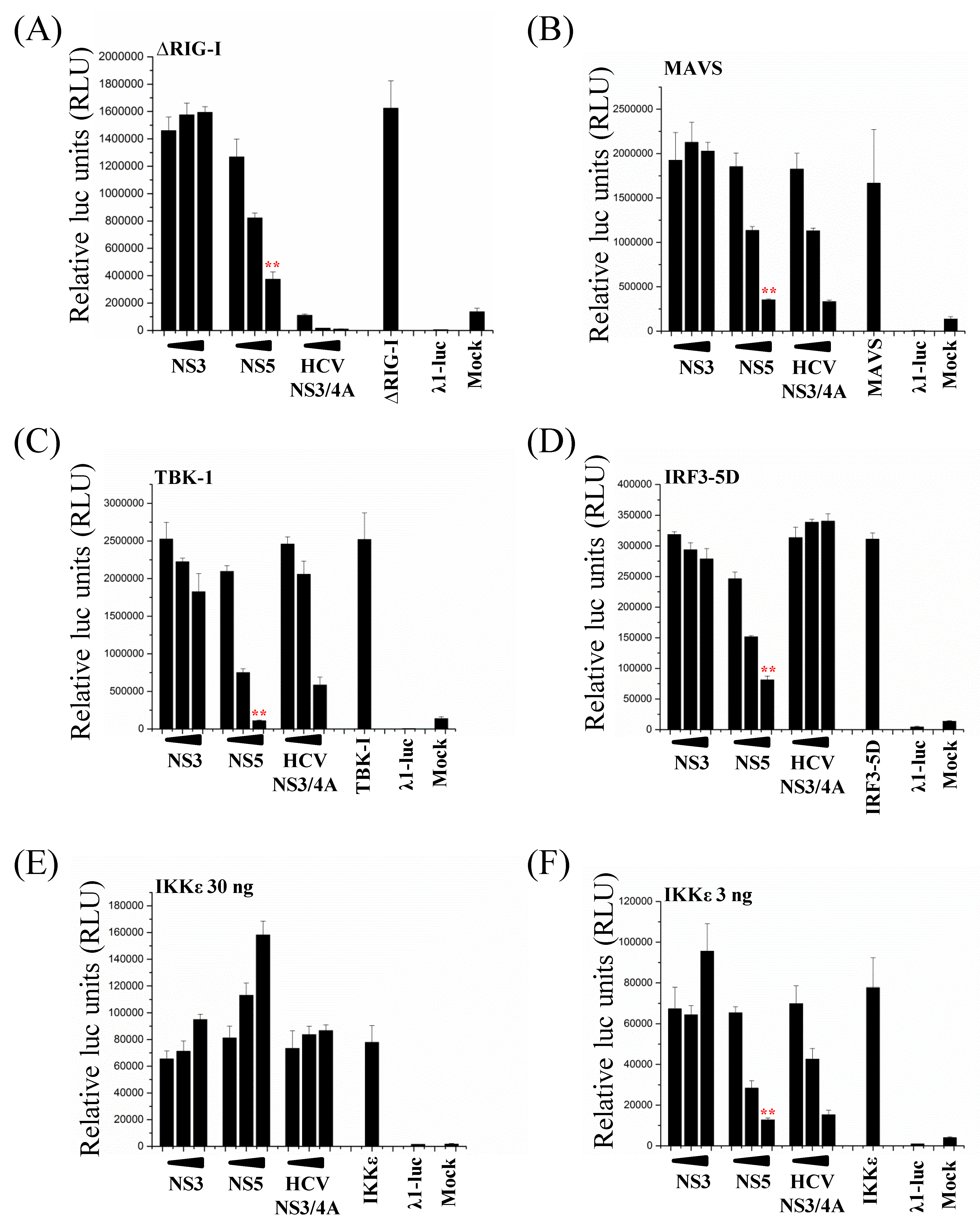
3.6. ZIKV NS5 Inhibits the RIG-I Pathway at Multiple Levels
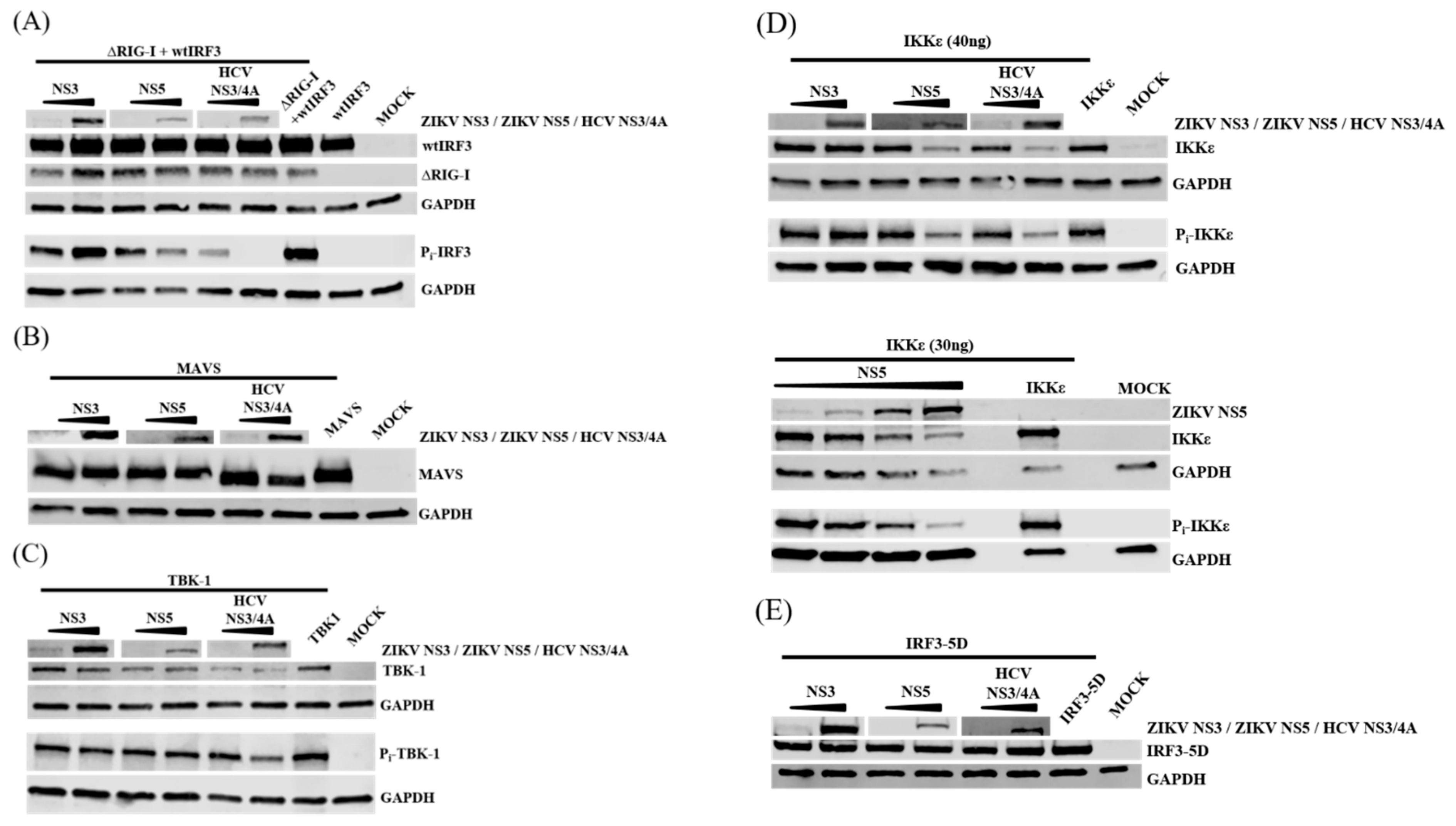
3.7. NS5 Inhibits Phosphorylation of IRF3 and IKKε
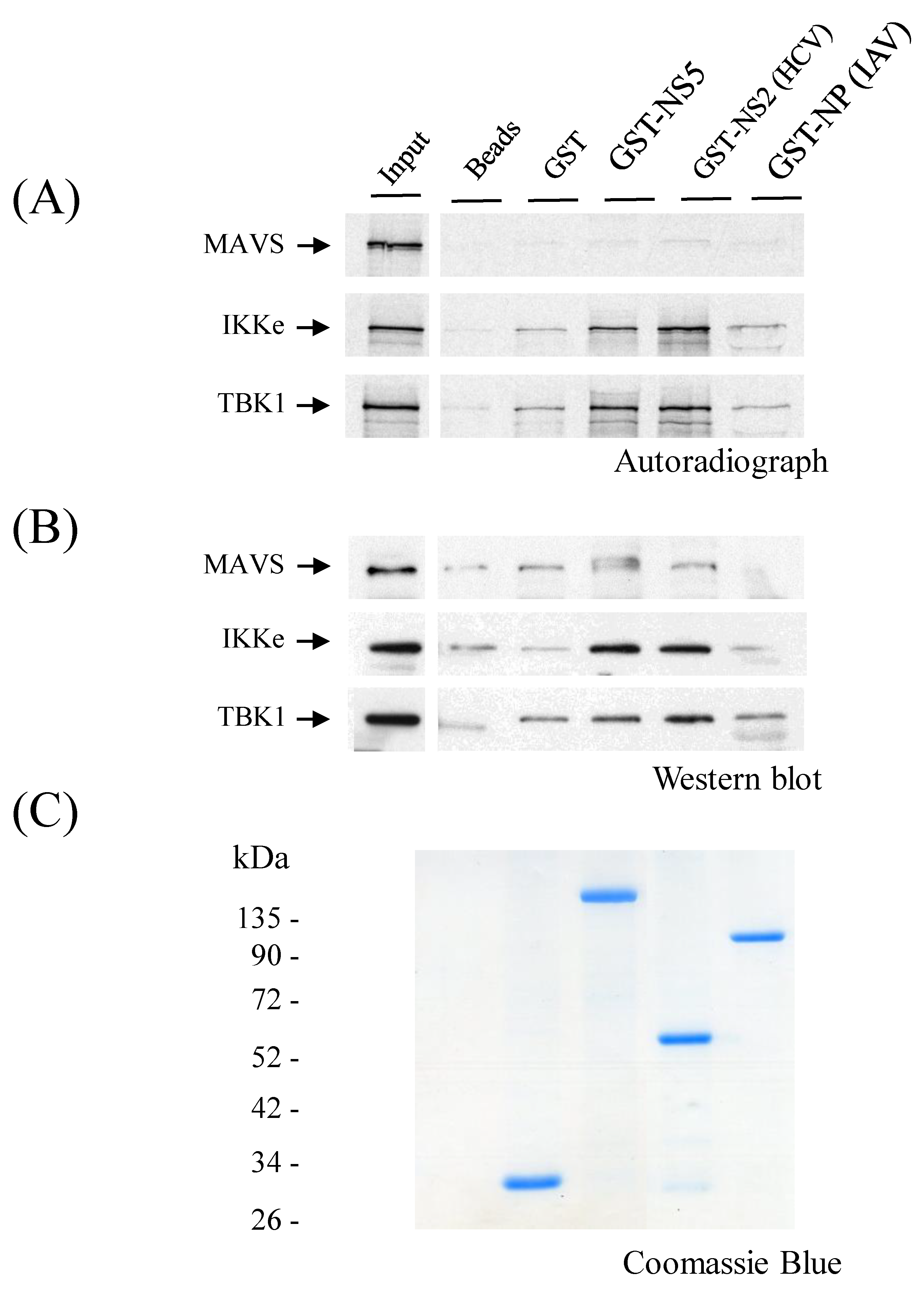
3.8. ZIKV NS5 Binds to IKKε
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Musso, D.; Gubler, D.J. Zika Virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef] [PubMed]
- Wikan, N.; Smith, D.R. Zika Virus: History of a Newly Emerging Arbovirus. Lancet Infect. Dis. 2016, 16, e119–e126. [Google Scholar] [CrossRef]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.N.; Strange, D.P.; Maharaj, P.N.; Shi, P.Y.; Verma, S. Zika Virus Infects Human Sertoli Cells and Modulates the Integrity of the in Vitro Blood-Testis Barrier Model. J. Virol. 2017, 91, e00623-17. [Google Scholar] [CrossRef] [PubMed]
- Tabata, T.; Petitt, M.; Puerta-Guardo, H.; Michlmayr, D.; Wang, C.; Fang-Hoover, J.; Harris, E.; Pereira, L. Zika Virus Targets Different Primary Human Placental Cells, Suggesting Two Routes for Vertical Transmission. Cell. Host Microbe 2016, 20, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Quicke, K.M.; Bowen, J.R.; Johnson, E.L.; McDonald, C.E.; Ma, H.; O’Neal, J.T.; Rajakumar, A.; Wrammert, J.; Rimawi, B.H.; Pulendran, B.; et al. Zika Virus Infects Human Placental Macrophages. Cell. Host Microbe 2016, 20, 83–90. [Google Scholar] [CrossRef]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates their Growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Onorati, M.; Li, Z.; Liu, F.; Sousa, A.M.M.; Nakagawa, N.; Li, M.; Dell’Anno, M.T.; Gulden, F.O.; Pochareddy, S.; Tebbenkamp, A.T.N.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016, 16, 2576–2592. [Google Scholar] [CrossRef]
- Hemann, E.A.; Gale, M., Jr.; Savan, R. Interferon Lambda Genetics and Biology in Regulation of Viral Control. Front. Immunol. 2017, 8, 1707. [Google Scholar] [CrossRef]
- Vladimer, G.I.; Gorna, M.W.; Superti-Furga, G. IFITs: Emerging Roles as Key Anti-Viral Proteins. Front. Immunol. 2014, 5, 94. [Google Scholar] [CrossRef]
- Fitzgerald, K.A. The Interferon Inducible Gene: Viperin. J. Interferon Cytokine Res. 2011, 31, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Kochs, G. Human MxA Protein: An Interferon-Induced Dynamin-Like GTPase with Broad Antiviral Activity. J. Interferon Cytokine Res. 2011, 31, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Vignali, D.A. STAT Heterodimers in Immunity: A Mixed Message or a Unique Signal? Jak-Stat 2013, 2, e23060. [Google Scholar] [CrossRef] [PubMed]
- Bowen, J.R.; Zimmerman, M.G.; Suthar, M.S. Taking the Defensive: Immune Control of Zika Virus Infection. Virus Res. 2017, 254, 21–26. [Google Scholar] [CrossRef]
- Hertzog, J.; Dias Junior, A.G.; Rigby, R.E.; Donald, C.L.; Mayer, A.; Sezgin, E.; Song, C.; Jin, B.; Hublitz, P.; Eggeling, C.; et al. Infection with a Brazilian Isolate of Zika Virus Generates RIG-I Stimulatory RNA and the Viral NS5 Protein Blocks Type I IFN Induction and Signaling. Eur. J. Immunol. 2018, 48, 1120–1136. [Google Scholar] [CrossRef]
- Bowen, J.R.; Quicke, K.M.; Maddur, M.S.; O’Neal, J.T.; McDonald, C.E.; Fedorova, N.B.; Puri, V.; Shabman, R.S.; Pulendran, B.; Suthar, M.S. Zika Virus Antagonizes Type I Interferon Responses during Infection of Human Dendritic Cells. PLoS Pathog. 2017, 13, e1006164. [Google Scholar] [CrossRef]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sanchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika Virus Inhibits Type-I Interferon Production and Downstream Signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef]
- Chaudhary, V.; Yuen, K.S.; Chan, J.F.; Chan, C.P.; Wang, P.H.; Cai, J.P.; Zhang, S.; Liang, M.; Kok, K.H.; Chan, C.P.; et al. Selective Activation of Type II Interferon Signaling by Zika Virus NS5 Protein. J. Virol. 2017, 91, e00163-17. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, Q.; Zhou, J.; Xie, W.; Chen, C.; Wang, Z.; Yang, H.; Cui, J. Zika Virus Evades Interferon-Mediated Antiviral Response through the Co-Operation of Multiple Nonstructural Proteins in Vitro. Cell Discov. 2017, 3, 17006. [Google Scholar] [CrossRef]
- Xia, H.; Luo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.; Vasconcelos, P.F.C.; et al. An Evolutionary NS1 Mutation Enhances Zika Virus Evasion of Host Interferon Induction. Nat. Commun. 2018, 9, 414. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yang, S.; He, J.; Guest, J.D.; Ma, Z.; Yang, L.; Pierce, B.G.; Tang, Q.; Zhang, Y.J. Zika Virus NS5 Protein Antagonizes Type I Interferon Production Via Blocking TBK1 Activation. Virology 2019, 527, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Munir, M.; Berg, M. The Multiple Faces of Proteinkinase R in Antiviral Defense. Virulence 2013, 4, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Feldherr, C.; Akin, D.; Moore, M.S. The Nuclear Import Factor p10 Regulates the Functional Size of the Nuclear Pore Complex during Oogenesis. J. Cell Sci. 1998, 111 Pt 13, 1889–1896. [Google Scholar]
- Fournier, E.; Moules, V.; Essere, B.; Paillart, J.C.; Sirbat, J.D.; Isel, C.; Cavalier, A.; Rolland, J.P.; Thomas, D.; Lina, B.; et al. A Supramolecular Assembly Formed by Influenza a Virus Genomic RNA Segments. Nucleic Acids Res. 2012, 40, 2197–2209. [Google Scholar] [CrossRef] [PubMed]
- Nabi, I.R.; Le, P.U. Caveolae/Raft-Dependent Endocytosis. J. Cell Biol. 2003, 161, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.H. Viral Encounters with 2’,5’-Oligoadenylate Synthetase and RNase L during the Interferon Antiviral Response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef]
- Sharma, S.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the Interferon Antiviral Response through an IKK-Related Pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef]
- Airenne, K.J.; Peltomaa, E.; Hytonen, V.P.; Laitinen, O.H.; Yla-Herttuala, S. Improved Generation of Recombinant Baculovirus Genomes in Escherichia Coli. Nucleic Acids Res. 2003, 31, e101. [Google Scholar] [CrossRef]
- Kaukinen, P.; Sillanpaa, M.; Nousiainen, L.; Melen, K.; Julkunen, I. Hepatitis C Virus NS2 Protease Inhibits Host Cell Antiviral Response by Inhibiting IKKepsilon and TBK1 Functions. J. Med. Virol. 2013, 85, 71–82. [Google Scholar] [CrossRef]
- Matikainen, S.; Siren, J.; Tissari, J.; Veckman, V.; Pirhonen, J.; Severa, M.; Sun, Q.; Lin, R.; Meri, S.; Uze, G.; et al. Tumor Necrosis Factor Alpha Enhances Influenza a Virus-Induced Expression of Antiviral Cytokines by Activating RIG-I Gene Expression. J. Virol. 2006, 80, 3515–3522. [Google Scholar] [CrossRef] [PubMed]
- Melen, K.; Kakkola, L.; He, F.; Airenne, K.; Vapalahti, O.; Karlberg, H.; Mirazimi, A.; Julkunen, I. Production, Purification and Immunogenicity of Recombinant Ebola Virus Proteins—A Comparison of Freund’s Adjuvant and Adjuvant System 03. J. Virol. Methods 2017, 242, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, H.; Taketa, K.; Miyano, K.; Yamane, T.; Sato, J. Growth of Human Hepatoma Cells Lines with Differentiated Functions in Chemically Defined Medium. Cancer Res. 1982, 42, 3858–3863. [Google Scholar] [PubMed]
- Summers, M.D.; Smith, G.E. A Manual of Methods for Baculovirus Vectors and Insect Cell Culture Procedures. Texas Agric. Exp. Stn. Bull. 1986, 1555, 1–57. [Google Scholar]
- Hagen, M.; Chung, T.D.; Butcher, J.A.; Krystal, M. Recombinant Influenza Virus Polymerase: Requirement of both 5’ and 3’ Viral Ends for Endonuclease Activity. J. Virol. 1994, 68, 1509–1515. [Google Scholar] [PubMed]
- Driggers, R.W.; Ho, C.Y.; Korhonen, E.M.; Kuivanen, S.; Jaaskelainen, A.J.; Smura, T.; Rosenberg, A.; Hill, D.A.; DeBiasi, R.L.; Vezina, G.; et al. Zika Virus Infection with Prolonged Maternal Viremia and Fetal Brain Abnormalities. N. Engl. J. Med. 2016, 374, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Buhler, S.; Bartenschlager, R. The Non-Structural Protein 4A of Dengue Virus is an Integral Membrane Protein Inducing Membrane Alterations in a 2K-Regulated Manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef]
- Zmurko, J.; Neyts, J.; Dallmeier, K. Flaviviral NS4b, Chameleon and Jack-in-the-Box Roles in Viral Replication and Pathogenesis, and a Molecular Target for Antiviral Intervention. Rev. Med. Virol. 2015, 25, 205–223. [Google Scholar] [CrossRef]
- Zou, J.; Xie, X.; Wang, Q.Y.; Dong, H.; Lee, M.Y.; Kang, C.; Yuan, Z.; Shi, P.Y. Characterization of Dengue Virus NS4A and NS4B Protein Interaction. J. Virol. 2015, 89, 3455–3470. [Google Scholar] [CrossRef]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and Essential Roles of Transcription Factors IRF-3 and IRF-7 in Response to Viruses for IFN-Alpha/Beta Gene Induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef]
- Zhao, B.; Yi, G.; Du, F.; Chuang, Y.C.; Vaughan, R.C.; Sankaran, B.; Kao, C.C.; Li, P. Structure and Function of the Zika Virus Full-Length NS5 Protein. Nat. Commun. 2017, 8, 14762. [Google Scholar] [CrossRef] [PubMed]
- Osterlund, P.I.; Pietila, T.E.; Veckman, V.; Kotenko, S.V.; Julkunen, I. IFN Regulatory Factor Family Members Differentially Regulate the Expression of Type III IFN (IFN-Lambda) Genes. J. Immunol. 2007, 179, 3434–3442. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Osterlund, P.; Fagerlund, R.; Rios, D.N.; Hoffmann, A.; Poranen, M.M.; Bamford, D.H.; Julkunen, I. MAP Kinase p38alpha Regulates Type III Interferon (IFN-Lambda1) Gene Expression in Human Monocyte-Derived Dendritic Cells in Response to RNA Stimulation. J. Leukoc. Biol. 2015, 97, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Kaukinen, P.; Sillanpaa, M.; Kotenko, S.; Lin, R.; Hiscott, J.; Melen, K.; Julkunen, I. Hepatitis C Virus NS2 and NS3/4A Proteins are Potent Inhibitors of Host Cell Cytokine/Chemokine Gene Expression. Virol. J. 2006, 3, 66-422X-3-66. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.; Lennemann, N.J.; Ouyang, Y.; Bramley, J.C.; Morosky, S.; Marques, E.T., Jr.; Cherry, S.; Sadovsky, Y.; Coyne, C.B. Type III Interferons Produced by Human Placental Trophoblasts Confer Protection Against Zika Virus Infection. Cell Host Microbe 2016, 19, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Jagger, B.W.; Miner, J.J.; Cao, B.; Arora, N.; Smith, A.M.; Kovacs, A.; Mysorekar, I.U.; Coyne, C.B.; Diamond, M.S. Gestational Stage and IFN-Lambda Signaling Regulate ZIKV Infection in Utero. Cell Host Microbe 2017, 22, 366–376.e3. [Google Scholar] [CrossRef]
- Morrison, J.; Aguirre, S.; Fernandez-Sesma, A. Innate Immunity Evasion by Dengue Virus. Viruses 2012, 4, 397–413. [Google Scholar] [CrossRef]
- Esser-Nobis, K.; Aarreberg, L.D.; Roby, J.A.; Fairgrieve, M.R.; Green, R.; Gale, M., Jr. Comparative Analysis of African and Asian Lineage-Derived Zika Virus Strains Reveals Differences in Activation of and Sensitivity to Antiviral Innate Immunity. J. Virol. 2019, 93, e00640-19. [Google Scholar] [CrossRef]
- Morrison, J.; Laurent-Rolle, M.; Maestre, A.M.; Rajsbaum, R.; Pisanelli, G.; Simon, V.; Mulder, L.C.; Fernandez-Sesma, A.; Garcia-Sastre, A. Dengue Virus Co-Opts UBR4 to Degrade STAT2 and Antagonize Type I Interferon Signaling. PLoS Pathog. 2013, 9, e1003265. [Google Scholar] [CrossRef]
- Lubick, K.J.; Robertson, S.J.; McNally, K.L.; Freedman, B.A.; Rasmussen, A.L.; Taylor, R.T.; Walts, A.D.; Tsuruda, S.; Sakai, M.; Ishizuka, M.; et al. Flavivirus Antagonism of Type I Interferon Signaling Reveals Prolidase as a Regulator of IFNAR1 Surface Expression. Cell Host Microbe 2015, 18, 61–74. [Google Scholar] [CrossRef]
- Laurent-Rolle, M.; Boer, E.F.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The NS5 Protein of the Virulent West Nile Virus NY99 Strain is a Potent Antagonist of Type I Interferon-Mediated JAK-STAT Signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Rolle, M.; Morrison, J.; Rajsbaum, R.; Macleod, J.M.L.; Pisanelli, G.; Pham, A.; Ayllon, J.; Miorin, L.; Martinez, C.; tenOever, B.R.; et al. The Interferon Signaling Antagonist Function of Yellow Fever Virus NS5 Protein is Activated by Type I Interferon. Cell Host Microbe 2014, 16, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Wang, X.J.; Clark, D.C.; Lobigs, M.; Hall, R.A.; Khromykh, A.A. A Single Amino Acid Substitution in the West Nile Virus Nonstructural Protein NS2A Disables its Ability to Inhibit Alpha/Beta Interferon Induction and Attenuates Virus Virulence in Mice. J. Virol. 2006, 80, 2396–2404. [Google Scholar] [CrossRef]
- Anglero-Rodriguez, Y.I.; Pantoja, P.; Sariol, C.A. Dengue Virus Subverts the Interferon Induction Pathway Via NS2B/3 Protease-IkappaB Kinase Epsilon Interaction. Clin. Vaccine Immunol. 2014, 21, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ye, F.; Zhu, N.; Wang, W.; Deng, Y.; Zhao, Z.; Tan, W. Middle East Respiratory Syndrome Coronavirus ORF4b Protein Inhibits Type I Interferon Production through both Cytoplasmic and Nuclear Targets. Sci. Rep. 2015, 5, 17554. [Google Scholar] [CrossRef] [PubMed]
- Pythoud, C.; Rodrigo, W.W.; Pasqual, G.; Rothenberger, S.; Martinez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Arenavirus Nucleoprotein Targets Interferon Regulatory Factor-Activating Kinase IKKepsilon. J. Virol. 2012, 86, 7728–7738. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tan, X.F.; Thurmond, S.; Zhang, Z.M.; Lin, A.; Hai, R.; Song, J. The Structure of Zika Virus NS5 Reveals a Conserved Domain Conformation. Nat. Commun. 2017, 8, 14763. [Google Scholar] [CrossRef] [PubMed]
- Coutard, B.; Barral, K.; Lichiere, J.; Selisko, B.; Martin, B.; Aouadi, W.; Lombardia, M.O.; Debart, F.; Vasseur, J.J.; Guillemot, J.C.; et al. Zika Virus Methyltransferase: Structure and Functions for Drug Design Perspectives. J. Virol. 2017, 91, e02202-16. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lundberg, R.; Melén, K.; Westenius, V.; Jiang, M.; Österlund, P.; Khan, H.; Vapalahti, O.; Julkunen, I.; Kakkola, L. Zika Virus Non-Structural Protein NS5 Inhibits the RIG-I Pathway and Interferon Lambda 1 Promoter Activation by Targeting IKK Epsilon. Viruses 2019, 11, 1024. https://doi.org/10.3390/v11111024
Lundberg R, Melén K, Westenius V, Jiang M, Österlund P, Khan H, Vapalahti O, Julkunen I, Kakkola L. Zika Virus Non-Structural Protein NS5 Inhibits the RIG-I Pathway and Interferon Lambda 1 Promoter Activation by Targeting IKK Epsilon. Viruses. 2019; 11(11):1024. https://doi.org/10.3390/v11111024
Chicago/Turabian StyleLundberg, Rickard, Krister Melén, Veera Westenius, Miao Jiang, Pamela Österlund, Hira Khan, Olli Vapalahti, Ilkka Julkunen, and Laura Kakkola. 2019. "Zika Virus Non-Structural Protein NS5 Inhibits the RIG-I Pathway and Interferon Lambda 1 Promoter Activation by Targeting IKK Epsilon" Viruses 11, no. 11: 1024. https://doi.org/10.3390/v11111024
APA StyleLundberg, R., Melén, K., Westenius, V., Jiang, M., Österlund, P., Khan, H., Vapalahti, O., Julkunen, I., & Kakkola, L. (2019). Zika Virus Non-Structural Protein NS5 Inhibits the RIG-I Pathway and Interferon Lambda 1 Promoter Activation by Targeting IKK Epsilon. Viruses, 11(11), 1024. https://doi.org/10.3390/v11111024