Modeling Arboviral Infection in Mice Lacking the Interferon Alpha/Beta Receptor

 , ,
, ,
Abstract
1. Introduction
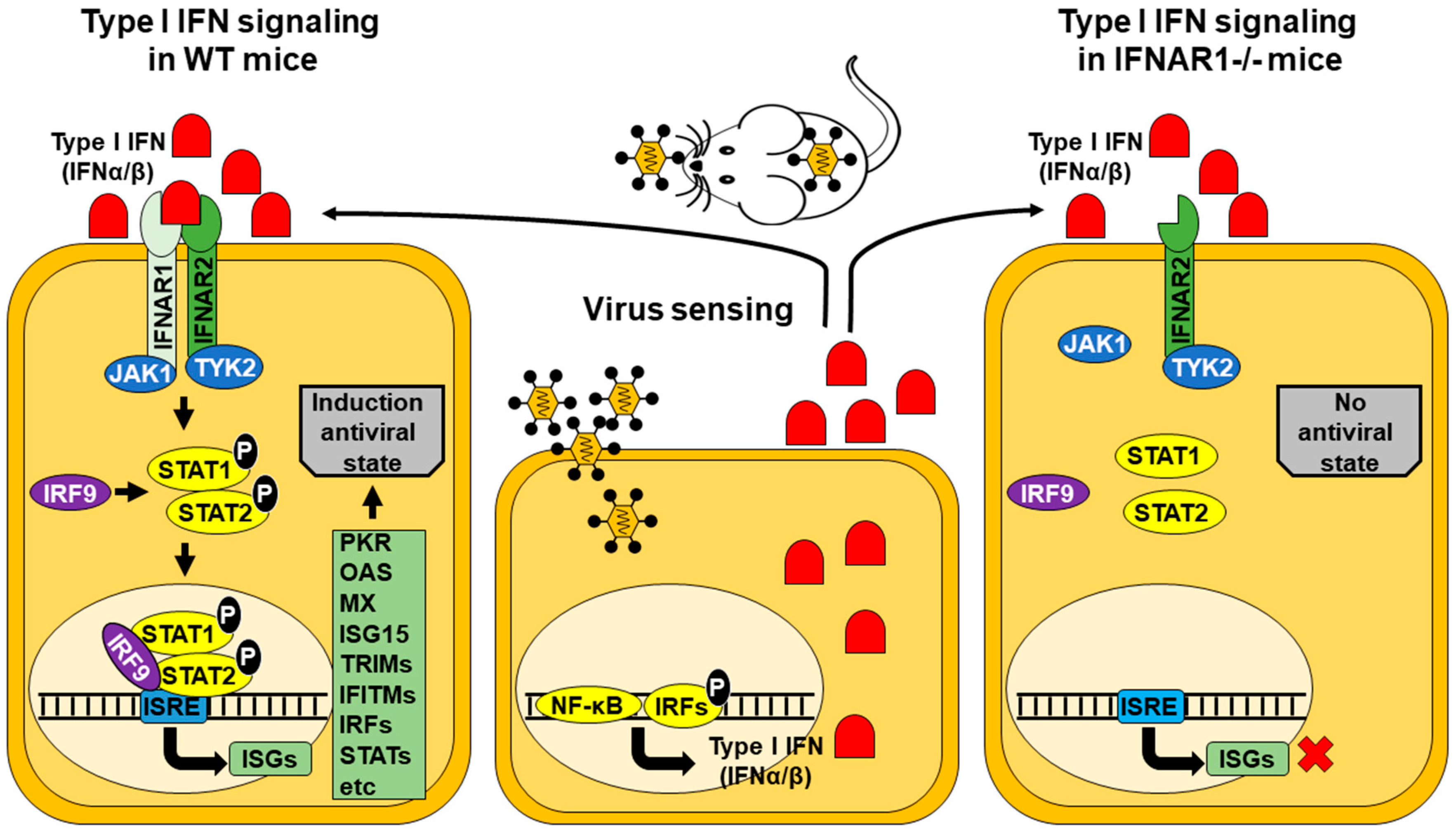
2. IFNAR(−/−) Mice
3. Families Included in the Order Bunyavirales
3.1. Rift Valley Fever Virus (Family Phenuiviridae)
3.2. Crimean Congo Fever Virus (Family Nairoviridae)
3.3. Schmallenberg Virus (Family Peribunyaviridae)
4. Family Flaviviridae
4.1. Dengue Virus
4.2. Yellow Fever Virus
4.3. Zika Virus
4.4. West Nile Virus and Japanese Encephalitis Virus
5. Family Togaviridae
5.1. Chikungunya Virus
5.2. Other Alphavirus
6. Family Rhabdoviridae
7. Family Orthomyxoviridae
8. Family Reoviridae
8.1. Bluetongue Virus
8.2. African Horse Sickness Virus and Epizootic Hemorrhagic Disease Virus
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pingen, M.; Schmid, M.A.; Harris, E.; McKimmie, C.S. Mosquito Biting Modulates Skin Response to Virus Infection. Trends Parasitol. 2017, 33, 645–657. [Google Scholar] [CrossRef]
- Hermance, M.E.; Thangamani, S. Tick Saliva Enhances Powassan Virus Transmission to the Host, Influencing Its Dissemination and the Course of Disease. J. Virol. 2015, 89, 7852–7860. [Google Scholar] [CrossRef]
- Pingen, M.; Bryden, S.R.; Pondeville, E.; Schnettler, E.; Kohl, A.; Merits, A.; Fazakerley, J.K.; Graham, G.J.; McKimmie, C.S. Host Inflammatory Response to Mosquito Bites Enhances the Severity of Arbovirus Infection. Immunity 2016, 44, 1455–1469. [Google Scholar] [CrossRef]
- Pages, N.; Breard, E.; Urien, C.; Talavera, S.; Viarouge, C.; Lorca-Oro, C.; Jouneau, L.; Charley, B.; Zientara, S.; Bensaid, A.; et al. Culicoides midge bites modulate the host response and impact on bluetongue virus infection in sheep. PLoS ONE 2014, 9, e83683. [Google Scholar] [CrossRef]
- Zivcec, M.; Safronetz, D.; Scott, D.; Robertson, S.; Ebihara, H.; Feldmann, H. Lethal Crimean-Congo hemorrhagic fever virus infection in interferon alpha/beta receptor knockout mice is associated with high viral loads, proinflammatory responses, and coagulopathy. J. Infect. Dis. 2013, 207, 1909–1921. [Google Scholar] [CrossRef]
- Calvo-Pinilla, E.; Rodriguez-Calvo, T.; Anguita, J.; Sevilla, N.; Ortego, J. Establishment of a bluetongue virus infection model in mice that are deficient in the alpha/beta interferon receptor. PLoS ONE 2009, 4, e5171. [Google Scholar] [CrossRef]
- Zhao, J.; Li, K.; Wohlford-Lenane, C.; Agnihothram, S.S.; Fett, C.; Gale, M.J., Jr.; Baric, R.S.; Enjuanes, L.; Gallagher, T.; McCray, P.B., Jr.; et al. Rapid generation of a mouse model for Middle East respiratory syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 4970–4975. [Google Scholar] [CrossRef] [PubMed]
- Bird, B.H.; Spengler, J.R.; Chakrabarti, A.K.; Khristova, M.L.; Sealy, T.K.; Coleman-McCray, J.D.; Martin, B.E.; Dodd, K.A.; Goldsmith, C.S.; Sanders, J.; et al. Humanized Mouse Model of Ebola Virus Disease Mimics the Immune Responses in Human Disease. J. Infect. Dis. 2016, 213, 703–711. [Google Scholar] [CrossRef]
- Gubareva, L.V.; McCullers, J.A.; Bethell, R.C.; Webster, R.G. Characterization of influenza A/HongKong/156/97 (H5N1) virus in a mouse model and protective effect of zanamivir on H5N1 infection in mice. J. Infect. Dis. 1998, 178, 1592–1596. [Google Scholar] [CrossRef]
- Subbarao, K.; McAuliffe, J.; Vogel, L.; Fahle, G.; Fischer, S.; Tatti, K.; Packard, M.; Shieh, W.J.; Zaki, S.; Murphy, B. Prior infection and passive transfer of neutralizing antibody prevent replication of severe acute respiratory syndrome coronavirus in the respiratory tract of mice. J. Virol. 2004, 78, 3572–3577. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.; Steele, K.E.; Shamblin, J.; Honko, A.; Johnson, J.; Reed, C.; Kennedy, M.; Chapman, J.L.; Hensley, L.E. The pathogenesis of Rift Valley fever virus in the mouse model. Virology 2010, 407, 256–267. [Google Scholar] [CrossRef]
- Muller, U.; Steinhoff, U.; Reis, L.F.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.M.; Aguet, M. Functional role of type I and type II interferons in antiviral defense. Science 1994, 264, 1918–1921. [Google Scholar] [CrossRef]
- Staeheli, P.; Danielson, P.; Haller, O.; Sutcliffe, J.G. Transcriptional activation of the mouse Mx gene by type I interferon. Mol. Cell. Biol. 1986, 6, 4770–4774. [Google Scholar] [CrossRef]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef]
- Lee, A.J.; Chen, B.; Chew, M.V.; Barra, N.G.; Shenouda, M.M.; Nham, T.; van Rooijen, N.; Jordana, M.; Mossman, K.L.; Schreiber, R.D.; et al. Inflammatory monocytes require type I interferon receptor signaling to activate NK cells via IL-18 during a mucosal viral infection. J. Exp. Med. 2017, 214, 1153–1167. [Google Scholar] [CrossRef]
- Lee, C.K.; Rao, D.T.; Gertner, R.; Gimeno, R.; Frey, A.B.; Levy, D.E. Distinct requirements for IFNs and STAT1 in NK cell function. J. Immunol. 2000, 165, 3571–3577. [Google Scholar] [CrossRef]
- Nguyen, K.B.; Salazar-Mather, T.P.; Dalod, M.Y.; Van Deusen, J.B.; Wei, X.Q.; Liew, F.Y.; Caligiuri, M.A.; Durbin, J.E.; Biron, C.A. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J. Immunol. 2002, 169, 4279–4287. [Google Scholar] [CrossRef]
- Le Bon, A.; Etchart, N.; Rossmann, C.; Ashton, M.; Hou, S.; Gewert, D.; Borrow, P.; Tough, D.F. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 2003, 4, 1009–1015. [Google Scholar] [CrossRef]
- Tanabe, Y.; Nishibori, T.; Su, L.; Arduini, R.M.; Baker, D.P.; David, M. Cutting edge: Role of STAT1, STAT3, and STAT5 in IFN-alpha beta responses in T lymphocytes. J. Immunol. 2005, 174, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Dondi, E.; Rogge, L.; Lutfalla, G.; Uze, G.; Pellegrini, S. Down-modulation of responses to type I IFN upon T cell activation. J. Immunol. 2003, 170, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Havenar-Daughton, C.; Kolumam, G.A.; Murali-Krishna, K. Cutting Edge: The direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J. Immunol. 2006, 176, 3315–3319. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef]
- Takaoka, A.; Hayakawa, S.; Yanai, H.; Stoiber, D.; Negishi, H.; Kikuchi, H.; Sasaki, S.; Imai, K.; Shibue, T.; Honda, K.; et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003, 424, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Kraus, J.; Ling, A.K.; Hamm, S.; Voigt, K.; Oschmann, P.; Engelhardt, B. Interferon-beta stabilizes barrier characteristics of brain endothelial cells in vitro. Ann. Neurol. 2004, 56, 192–205. [Google Scholar] [CrossRef]
- Markowitz, C.E. Interferon-beta: Mechanism of action and dosing issues. Neurology 2007, 68, S8–S11. [Google Scholar] [CrossRef]
- Daniels, B.P.; Holman, D.W.; Cruz-Orengo, L.; Jujjavarapu, H.; Durrant, D.M.; Klein, R.S. Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. mBio 2014, 5, e01476-14. [Google Scholar] [CrossRef]
- Bouloy, M.; Janzen, C.; Vialat, P.; Khun, H.; Pavlovic, J.; Huerre, M.; Haller, O. Genetic evidence for an interferon-antagonistic function of rift valley fever virus nonstructural protein NSs. J. Virol. 2001, 75, 1371–1377. [Google Scholar] [CrossRef]
- Lorenzo, G.; Martin-Folgar, R.; Hevia, E.; Boshra, H.; Brun, A. Protection against lethal Rift Valley fever virus (RVFV) infection in transgenic IFNAR(−/−) mice induced by different DNA vaccination regimens. Vaccine 2010, 28, 2937–2944. [Google Scholar] [CrossRef]
- Boshra, H.; Lorenzo, G.; Rodriguez, F.; Brun, A. A DNA vaccine encoding ubiquitinated Rift Valley fever virus nucleoprotein provides consistent immunity and protects IFNAR(−/−) mice upon lethal virus challenge. Vaccine 2011, 29, 4469–4475. [Google Scholar] [CrossRef] [PubMed]
- Borrego, B.; Lorenzo, G.; Mota-Morales, J.D.; Almanza-Reyes, H.; Mateos, F.; Lopez-Gil, E.; de la Losa, N.; Burmistrov, V.A.; Pestryakov, A.N.; Brun, A.; et al. Potential application of silver nanoparticles to control the infectivity of Rift Valley fever virus in vitro and in vivo. Nanomedicine 2016, 12, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gil, E.; Lorenzo, G.; Hevia, E.; Borrego, B.; Eiden, M.; Groschup, M.; Gilbert, S.C.; Brun, A. A single immunization with MVA expressing GnGc glycoproteins promotes epitope-specific CD8+-T cell activation and protects immune-competent mice against a lethal RVFV infection. PLoS Negl. Trop. Dis. 2013, 7, e2309. [Google Scholar] [CrossRef] [PubMed]
- Bereczky, S.; Lindegren, G.; Karlberg, H.; Akerstrom, S.; Klingstrom, J.; Mirazimi, A. Crimean-Congo hemorrhagic fever virus infection is lethal for adult type I interferon receptor-knockout mice. J. Gen. Virol. 2010, 91, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Zivcec, M.; Safronetz, D.; Scott, D.P.; Robertson, S.; Feldmann, H. Nucleocapsid protein-based vaccine provides protection in mice against lethal Crimean-Congo hemorrhagic fever virus challenge. PLoS Negl. Trop. Dis. 2018, 12, e0006628. [Google Scholar] [CrossRef]
- Canakoglu, N.; Berber, E.; Tonbak, S.; Ertek, M.; Sozdutmaz, I.; Aktas, M.; Kalkan, A.; Ozdarendeli, A. Immunization of knock-out alpha/beta interferon receptor mice against high lethal dose of Crimean-Congo hemorrhagic fever virus with a cell culture based vaccine. PLoS Negl. Trop. Dis. 2015, 9, e0003579. [Google Scholar] [CrossRef]
- Garrison, A.R.; Shoemaker, C.J.; Golden, J.W.; Fitzpatrick, C.J.; Suschak, J.J.; Richards, M.J.; Badger, C.V.; Six, C.M.; Martin, J.D.; Hannaman, D.; et al. A DNA vaccine for Crimean-Congo hemorrhagic fever protects against disease and death in two lethal mouse models. PLoS Negl. Trop. Dis. 2017, 11, e0005908. [Google Scholar] [CrossRef]
- Hinkula, J.; Devignot, S.; Akerstrom, S.; Karlberg, H.; Wattrang, E.; Bereczky, S.; Mousavi-Jazi, M.; Risinger, C.; Lindegren, G.; Vernersson, C.; et al. Immunization with DNA Plasmids Coding for Crimean-Congo Hemorrhagic Fever Virus Capsid and Envelope Proteins and/or Virus-Like Particles Induces Protection and Survival in Challenged Mice. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Buttigieg, K.R.; Dowall, S.D.; Findlay-Wilson, S.; Miloszewska, A.; Rayner, E.; Hewson, R.; Carroll, M.W. A novel vaccine against Crimean-Congo Haemorrhagic Fever protects 100% of animals against lethal challenge in a mouse model. PLoS ONE 2014, 9, e91516. [Google Scholar] [CrossRef]
- Oestereich, L.; Rieger, T.; Neumann, M.; Bernreuther, C.; Lehmann, M.; Krasemann, S.; Wurr, S.; Emmerich, P.; de Lamballerie, X.; Olschlager, S.; et al. Evaluation of antiviral efficacy of ribavirin, arbidol, and T-705 (favipiravir) in a mouse model for Crimean-Congo hemorrhagic fever. PLoS Negl. Trop. Dis. 2014, 8, e2804. [Google Scholar] [CrossRef]
- Kraatz, F.; Wernike, K.; Hechinger, S.; Konig, P.; Granzow, H.; Reimann, I.; Beer, M. Deletion mutants of Schmallenberg virus are avirulent and protect from virus challenge. J. Virol. 2015, 89, 1825–1837. [Google Scholar] [CrossRef] [PubMed]
- Boshra, H.Y.; Charro, D.; Lorenzo, G.; Sanchez, I.; Lazaro, B.; Brun, A.; Abrescia, N.G. DNA vaccination regimes against Schmallenberg virus infection in IFNAR(−/−) mice suggest two targets for immunization. Antivir. Res. 2017, 141, 107–115. [Google Scholar] [CrossRef]
- Wernike, K.; Aebischer, A.; Roman-Sosa, G.; Beer, M. The N-terminal domain of Schmallenberg virus envelope protein Gc is highly immunogenic and can provide protection from infection. Sci. Rep. 2017, 7, 42500. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, V.V.; Milligan, G.N.; Bourne, N.; Barrett, A.D. Mouse models of dengue virus infection for vaccine testing. Vaccine 2015, 33, 7051–7060. [Google Scholar] [CrossRef] [PubMed]
- Prestwood, T.R.; Morar, M.M.; Zellweger, R.M.; Miller, R.; May, M.M.; Yauch, L.E.; Lada, S.M.; Shresta, S. Gamma interferon (IFN-gamma) receptor restricts systemic dengue virus replication and prevents paralysis in IFN-alpha/beta receptor-deficient mice. J. Virol. 2012, 86, 12561–12570. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B. Neutralization and antibody-dependent enhancement of dengue viruses. Adv. Virus Res. 2003, 60, 421–467. [Google Scholar]
- Halstead, S.B. Dengue. Lancet 2007, 370, 1644–1652. [Google Scholar] [CrossRef]
- Orozco, S.; Schmid, M.A.; Parameswaran, P.; Lachica, R.; Henn, M.R.; Beatty, R.; Harris, E. Characterization of a model of lethal dengue virus 2 infection in C57BL/6 mice deficient in the alpha/beta interferon receptor. J. Gen. Virol. 2012, 93, 2152–2157. [Google Scholar] [CrossRef]
- Meier, K.C.; Gardner, C.L.; Khoretonenko, M.V.; Klimstra, W.B.; Ryman, K.D. A mouse model for studying viscerotropic disease caused by yellow fever virus infection. PLoS Pathog. 2009, 5, e1000614. [Google Scholar] [CrossRef]
- Dowall, S.D.; Graham, V.A.; Rayner, E.; Atkinson, B.; Hall, G.; Watson, R.J.; Bosworth, A.; Bonney, L.C.; Kitchen, S.; Hewson, R. A Susceptible Mouse Model for Zika Virus Infection. PLoS Negl. Trop. Dis. 2016, 10, e0004658. [Google Scholar] [CrossRef]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.L.; Tesh, R.B.; Azar, S.R.; Muruato, A.E.; Hanley, K.A.; Auguste, A.J.; Langsjoen, R.M.; Paessler, S.; Vasilakis, N.; Weaver, S.C. Characterization of a Novel Murine Model to Study Zika Virus. Am. J. Trop. Med. Hyg. 2016, 94, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Marzi, A.; Emanuel, J.; Callison, J.; McNally, K.L.; Arndt, N.; Chadinha, S.; Martellaro, C.; Rosenke, R.; Scott, D.P.; Safronetz, D.; et al. Lethal Zika Virus Disease Models in Young and Older Interferon alpha/beta Receptor Knock Out Mice. Front. Cell. Infect. Microbiol. 2018, 8, 117. [Google Scholar] [CrossRef]
- Perez, P.; Marín, Q.M.; Lazaro-Frias, A.; Jimenez de Oya, N.; Blazquez, A.B.; Escribano-Romero, E.; Sorzano, C.O.S.; Ortego, J.; Saiz, J.C.; Esteban, M.; et al. A Vaccine Based on a Modified Vaccinia Virus Ankara Vector Expressing Zika Virus Structural Proteins Controls Zika Virus Replication in Mice. Sci. Rep. 2018, 8, 17385. [Google Scholar] [CrossRef]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sanchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.A.; Diamond, M.S. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar] [CrossRef]
- Aoki, K.; Shimada, S.; Simantini, D.S.; Tun, M.M.; Buerano, C.C.; Morita, K.; Hayasaka, D. Type-I interferon response affects an inoculation dose-independent mortality in mice following Japanese encephalitis virus infection. Virol. J. 2014, 11, 105. [Google Scholar] [CrossRef]
- Couderc, T.; Chretien, F.; Schilte, C.; Disson, O.; Brigitte, M.; Guivel-Benhassine, F.; Touret, Y.; Barau, G.; Cayet, N.; Schuffenecker, I.; et al. A mouse model for Chikungunya: Young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 2008, 4, e29. [Google Scholar] [CrossRef]
- Ryman, K.D.; Klimstra, W.B.; Nguyen, K.B.; Biron, C.A.; Johnston, R.E. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J. Virol. 2000, 74, 3366–3378. [Google Scholar] [CrossRef]
- Schoneboom, B.A.; Lee, J.S.; Grieder, F.B. Early expression of IFN-alpha/beta and iNOS in the brains of Venezuelan equine encephalitis virus-infected mice. J. Interferon Cytokine Res. 2000, 20, 205–215. [Google Scholar] [CrossRef]
- Detje, C.N.; Meyer, T.; Schmidt, H.; Kreuz, D.; Rose, J.K.; Bechmann, I.; Prinz, M.; Kalinke, U. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J. Immunol. 2009, 182, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Wetzel, J.L.; Ramachandran, S.; Ogino, T.; Stohlman, S.A.; Bergmann, C.C.; Diamond, M.S.; Virgin, H.W.; Sen, G.C. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog. 2012, 8, e1002712. [Google Scholar] [CrossRef] [PubMed]
- Kochs, G.; Anzaghe, M.; Kronhart, S.; Wagner, V.; Gogesch, P.; Scheu, S.; Lienenklaus, S.; Waibler, Z. In Vivo Conditions Enable IFNAR-Independent Type I Interferon Production by Peritoneal CD11b+ Cells upon Thogoto Virus Infection. J. Virol. 2016, 90, 9330–9337. [Google Scholar] [CrossRef] [PubMed]
- Kochs, G.; Bauer, S.; Vogt, C.; Frenz, T.; Tschopp, J.; Kalinke, U.; Waibler, Z. Thogoto virus infection induces sustained type I interferon responses that depend on RIG-I-like helicase signaling of conventional dendritic cells. J. Virol. 2010, 84, 12344–12350. [Google Scholar] [CrossRef]
- Maclachlan, N.J.; Drew, C.P.; Darpel, K.E.; Worwa, G. The pathology and pathogenesis of bluetongue. J. Comp. Pathol. 2009, 141, 1–16. [Google Scholar] [CrossRef]
- Marin-Lopez, A.; Bermudez, R.; Calvo-Pinilla, E.; Moreno, S.; Brun, A.; Ortego, J. Pathological Characterization of IFNAR(−/−) Mice Infected with Bluetongue Virus Serotype 4. Int. J. Biol. Sci. 2016, 12, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Olivares, J.; Calvo-Pinilla, E.; Casanova, I.; Bachanek-Bankowska, K.; Chiam, R.; Maan, S.; Nieto, J.M.; Ortego, J.; Mertens, P.P. A modified vaccinia Ankara virus (MVA) vaccine expressing African horse sickness virus (AHSV) VP2 protects against AHSV challenge in an IFNAR −/− mouse model. PLoS ONE 2011, 6, e16503. [Google Scholar] [CrossRef]
- De la Poza, F.; Calvo-Pinilla, E.; Lopez-Gil, E.; Marin-Lopez, A.; Mateos, F.; Castillo-Olivares, J.; Lorenzo, G.; Ortego, J. Ns1 is a key protein in the vaccine composition to protect IFNAR(−/−) mice against infection with multiple serotypes of African horse sickness virus. PLoS ONE 2013, 8, e70197. [Google Scholar] [CrossRef] [PubMed]
- Eschbaumer, M.; Keller, M.; Beer, M.; Hoffmann, B. Epizootic hemorrhagic disease virus infection of type I interferon receptor deficient mice. Vet. Microbiol. 2012, 155, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Davies, F.G.; Martin, V. Recognizing Rift Valley Fever. Vet. Ital. 2006, 42, 31–53. [Google Scholar] [PubMed]
- Madani, T.A.; Al-Mazrou, Y.Y.; Al-Jeffri, M.H.; Mishkhas, A.A.; Al-Rabeah, A.M.; Turkistani, A.M.; Al-Sayed, M.O.; Abodahish, A.A.; Khan, A.S.; Ksiazek, T.G.; et al. Rift Valley fever epidemic in Saudi Arabia: Epidemiological, clinical, and laboratory characteristics. Clin. Infect. Dis. 2003, 37, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Mims, C.A. Rift Valley Fever virus in mice. I. General features of the infection. Br. J. Exp. Pathol. 1956, 37, 99–109. [Google Scholar] [PubMed]
- Whitehouse, C.A. Crimean-Congo hemorrhagic fever. Antivir. Res. 2004, 64, 145–160. [Google Scholar] [CrossRef]
- Wernike, K.; Beer, M. Schmallenberg Virus: A Novel Virus of Veterinary Importance. Adv. Virus Res. 2017, 99, 39–60. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, H.; Hoffmann, B.; Turan, N.; Cizmecigil, U.Y.; Richt, J.A.; Van der Poel, W.H. Detection and partial sequencing of Schmallenberg virus in cattle and sheep in Turkey. Vector Borne Zoonotic Dis. 2014, 14, 223–225. [Google Scholar] [CrossRef]
- Kurogi, H.; Inaba, Y.; Takahashi, E.; Sato, K.; Omori, T.; Miura, Y.; Goto, Y.; Fujiwara, Y.; Hatano, Y.; Kodama, K.; et al. Epizootic congenital arthrogryposis-hydranencephaly syndrome in cattle: Isolation of Akabane virus from affected fetuses. Arch. Virol. 1976, 51, 67–74. [Google Scholar] [CrossRef]
- Varela, M.; Schnettler, E.; Caporale, M.; Murgia, C.; Barry, G.; McFarlane, M.; McGregor, E.; Piras, I.M.; Shaw, A.; Lamm, C.; et al. Schmallenberg virus pathogenesis, tropism and interaction with the innate immune system of the host. PLoS Pathog. 2013, 9, e1003133. [Google Scholar] [CrossRef]
- Tauscher, K.; Wernike, K.; Fischer, M.; Wegelt, A.; Hoffmann, B.; Teifke, J.P.; Beer, M. Characterization of Simbu serogroup virus infections in type I interferon receptor knock-out mice. Arch. Virol. 2017, 162, 3119–3129. [Google Scholar] [CrossRef]
- Boyd, A.; Fazakerley, J.K.; Bridgen, A. Pathogenesis of Dugbe virus infection in wild-type and interferon-deficient mice. J. Gen. Virol. 2006, 87, 2005–2009. [Google Scholar] [CrossRef]
- Weber, F.; Bridgen, A.; Fazakerley, J.K.; Streitenfeld, H.; Kessler, N.; Randall, R.E.; Elliott, R.M. Bunyamwera bunyavirus nonstructural protein NSs counteracts the induction of alpha/beta interferon. J. Virol. 2002, 76, 7949–7955. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, B.; Paessler, S.; Walker, D.H.; Tesh, R.B.; Yu, X.J. The pathogenesis of severe fever with thrombocytopenia syndrome virus infection in alpha/beta interferon knockout mice: Insights into the pathologic mechanisms of a new viral hemorrhagic fever. J. Virol. 2014, 88, 1781–1786. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, D.; de Queiroz Prado, R.; Almeida Xavier, E.; Cristina de Oliveira, N.; da Matta Guedes, P.M.; da Silva, J.S.; Moraes Figueiredo, L.T.; Aquino, V.H. Immunocompetent mice model for dengue virus infection. Sci. World J. 2012, 2012, 525947. [Google Scholar] [CrossRef] [PubMed]
- Yauch, L.E.; Zellweger, R.M.; Kotturi, M.F.; Qutubuddin, A.; Sidney, J.; Peters, B.; Prestwood, T.R.; Sette, A.; Shresta, S. A protective role for dengue virus-specific CD8+ T cells. J. Immunol. 2009, 182, 4865–4873. [Google Scholar] [CrossRef] [PubMed]
- Zust, R.; Dong, H.; Li, X.F.; Chang, D.C.; Zhang, B.; Balakrishnan, T.; Toh, Y.X.; Jiang, T.; Li, S.H.; Deng, Y.Q.; et al. Rational design of a live attenuated dengue vaccine: 2′-o-methyltransferase mutants are highly attenuated and immunogenic in mice and macaques. PLoS Pathog. 2013, 9, e1003521. [Google Scholar] [CrossRef] [PubMed]
- Douam, F.; Ploss, A. Yellow Fever Virus: Knowledge Gaps Impeding the Fight Against an Old Foe. Trends Microbiol. 2018, 26, 913–928. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Rolle, M.; Morrison, J.; Rajsbaum, R.; Macleod, J.M.L.; Pisanelli, G.; Pham, A.; Ayllon, J.; Miorin, L.; Martinez, C.; tenOever, B.R.; et al. The interferon signaling antagonist function of yellow fever virus NS5 protein is activated by type I interferon. Cell Host Microbe 2014, 16, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.R.; Chen, T.H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lormeau, V.M.; Roche, C.; Teissier, A.; Robin, E.; Berry, A.L.; Mallet, H.P.; Sall, A.A.; Musso, D. Zika virus, French polynesia, South pacific, 2013. Emerg. Infect. Dis. 2014, 20, 1085–1086. [Google Scholar] [CrossRef]
- Tognarelli, J.; Ulloa, S.; Villagra, E.; Lagos, J.; Aguayo, C.; Fasce, R.; Parra, B.; Mora, J.; Becerra, N.; Lagos, N.; et al. A report on the outbreak of Zika virus on Easter Island, South Pacific, 2014. Arch. Virol. 2016, 161, 665–668. [Google Scholar] [CrossRef]
- Wong, G.; Qiu, X.G. Type I interferon receptor knockout mice as models for infection of highly pathogenic viruses with outbreak potential. Zool. Res. 2018, 39, 3–14. [Google Scholar] [CrossRef]
- Kindhauser, M.K.; Allen, T.; Frank, V.; Santhana, R.S.; Dye, C. Zika: The origin and spread of a mosquito-borne virus. Bull. World Health Organ. 2016, 94, 675. [Google Scholar] [CrossRef] [PubMed]
- Song, B.H.; Yun, S.I.; Woolley, M.; Lee, Y.M. Zika virus: History, epidemiology, transmission, and clinical presentation. J. Neuroimmunol. 2017, 308, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.I. Zika Virus Infection in Man. Trans. R. Soc. Trop. Med. Hyg. 1964, 58, 335–338. [Google Scholar] [CrossRef]
- Boeuf, P.; Drummer, H.E.; Richards, J.S.; Scoullar, M.J.; Beeson, J.G. The global threat of Zika virus to pregnancy: Epidemiology, clinical perspectives, mechanisms, and impact. BMC Med. 2016, 14, 112. [Google Scholar] [CrossRef] [PubMed]
- Frontera, J.A.; da Silva, I.R. Zika Getting on Your Nerves? The Association with the Guillain-Barre Syndrome. N. Engl. J. Med. 2016, 375, 1581–1582. [Google Scholar] [CrossRef] [PubMed]
- Deckard, D.T.; Chung, W.M.; Brooks, J.T.; Smith, J.C.; Woldai, S.; Hennessey, M.; Kwit, N.; Mead, P. Male-to-Male Sexual Transmission of Zika Virus—Texas, January 2016. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 372–374. [Google Scholar] [CrossRef]
- D’Ortenzio, E.; Matheron, S.; Yazdanpanah, Y.; de Lamballerie, X.; Hubert, B.; Piorkowski, G.; Maquart, M.; Descamps, D.; Damond, F.; Leparc-Goffart, I. Evidence of Sexual Transmission of Zika Virus. N. Engl. J. Med. 2016, 374, 2195–2198. [Google Scholar] [CrossRef]
- Hills, S.L.; Russell, K.; Hennessey, M.; Williams, C.; Oster, A.M.; Fischer, M.; Mead, P. Transmission of Zika Virus Through Sexual Contact with Travelers to Areas of Ongoing Transmission—Continental United States, 2016. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 215–216. [Google Scholar] [CrossRef]
- Davidson, A.; Slavinski, S.; Komoto, K.; Rakeman, J.; Weiss, D. Suspected Female-to-Male Sexual Transmission of Zika Virus—New York City, 2016. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 716–717. [Google Scholar] [CrossRef]
- Uraki, R.; Hwang, J.; Jurado, K.A.; Householder, S.; Yockey, L.J.; Hastings, A.K.; Homer, R.J.; Iwasaki, A.; Fikrig, E. Zika virus causes testicular atrophy. Sci. Adv. 2017, 3, e1602899. [Google Scholar] [CrossRef]
- Ma, W.; Li, S.; Ma, S.; Jia, L.; Zhang, F.; Zhang, Y.; Zhang, J.; Wong, G.; Zhang, S.; Lu, X.; et al. Zika Virus Causes Testis Damage and Leads to Male Infertility in Mice. Cell 2017, 168, 542. [Google Scholar] [CrossRef] [PubMed]
- Yockey, L.J.; Varela, L.; Rakib, T.; Khoury-Hanold, W.; Fink, S.L.; Stutz, B.; Szigeti-Buck, K.; Van den Pol, A.; Lindenbach, B.D.; Horvath, T.L.; et al. Vaginal Exposure to Zika Virus during Pregnancy Leads to Fetal Brain Infection. Cell 2016, 166, 1247–1256. [Google Scholar] [CrossRef]
- Yockey, L.J.; Jurado, K.A.; Arora, N.; Millet, A.; Rakib, T.; Milano, K.M.; Hastings, A.K.; Fikrig, E.; Kong, Y.; Horvath, T.L.; et al. Type I interferons instigate fetal demise after Zika virus infection. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, J.; Callison, J.; Dowd, K.A.; Pierson, T.C.; Feldmann, H.; Marzi, A. A VSV-based Zika virus vaccine protects mice from lethal challenge. Sci. Rep. 2018, 8, 11043. [Google Scholar] [CrossRef] [PubMed]
- Prow, N.A.; Liu, L.; Nakayama, E.; Cooper, T.H.; Yan, K.; Eldi, P.; Hazlewood, J.E.; Tang, B.; Le, T.T.; Setoh, Y.X.; et al. A vaccinia-based single vector construct multi-pathogen vaccine protects against both Zika and chikungunya viruses. Nat. Commun. 2018, 9, 1230. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Guo, X.; Shen, C.; Hao, X.; Sun, P.; Li, P.; Xu, T.; Hu, C.; Rose, O.; Zhou, H.; et al. Salivary factor LTRIN from Aedes aegypti facilitates the transmission of Zika virus by interfering with the lymphotoxin-beta receptor. Nat. Immunol. 2018, 19, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Colpitts, T.M.; Conway, M.J.; Montgomery, R.R.; Fikrig, E. West Nile Virus: Biology, transmission, and human infection. Clin. Microbiol. Rev. 2012, 25, 635–648. [Google Scholar] [CrossRef]
- Chen, C.C.; Jenkins, E.; Epp, T.; Waldner, C.; Curry, P.S.; Soos, C. Climate change and West Nile virus in a highly endemic region of North America. Int. J. Environ. Res. Public Health 2013, 10, 3052–3071. [Google Scholar] [CrossRef]
- Chancey, C.; Grinev, A.; Volkova, E.; Rios, M. The global ecology and epidemiology of West Nile virus. Biomed. Res. Int. 2015, 2015, 376230. [Google Scholar] [CrossRef]
- Keller, B.C.; Fredericksen, B.L.; Samuel, M.A.; Mock, R.E.; Mason, P.W.; Diamond, M.S.; Gale, M., Jr. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J. Virol. 2006, 80, 9424–9434. [Google Scholar] [CrossRef]
- Winkelmann, E.R.; Widman, D.G.; Xia, J.; Ishikawa, T.; Miller-Kittrell, M.; Nelson, M.H.; Bourne, N.; Scholle, F.; Mason, P.W.; Milligan, G.N. Intrinsic adjuvanting of a novel single-cycle flavivirus vaccine in the absence of type I interferon receptor signaling. Vaccine 2012, 30, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Erlanger, T.E.; Weiss, S.; Keiser, J.; Utzinger, J.; Wiedenmayer, K. Past, present, and future of Japanese encephalitis. Emerg. Infect. Dis. 2009, 15, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Basu, A. Japanese encephalitis—A pathological and clinical perspective. PLoS Negl. Trop. Dis. 2009, 3, e437. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.; Finsterbusch, K.; Lindquist, R.; Nair, S.; Lienenklaus, S.; Gekara, N.O.; Janik, D.; Weiss, S.; Kalinke, U.; Overby, A.K.; et al. Type I interferon protects mice from fatal neurotropic infection with Langat virus by systemic and local antiviral responses. J. Virol. 2014, 88, 12202–12212. [Google Scholar] [CrossRef]
- Lindqvist, R.; Upadhyay, A.; Overby, A.K. Tick-Borne Flaviviruses and the Type I Interferon Response. Viruses 2018, 10, 340. [Google Scholar] [CrossRef]
- Saxton-Shaw, K.D.; Ledermann, J.P.; Kenney, J.L.; Berl, E.; Graham, A.C.; Russo, J.M.; Powers, A.M.; Mutebi, J.P. The first outbreak of eastern equine encephalitis in Vermont: Outbreak description and phylogenetic relationships of the virus isolate. PLoS ONE 2015, 10, e0128712. [Google Scholar] [CrossRef]
- Adams, A.P.; Navarro-Lopez, R.; Ramirez-Aguilar, F.J.; Lopez-Gonzalez, I.; Leal, G.; Flores-Mayorga, J.M.; Travassos da Rosa, A.P.; Saxton-Shaw, K.D.; Singh, A.J.; Borland, E.M.; et al. Venezuelan equine encephalitis virus activity in the Gulf Coast region of Mexico, 2003–2010. PLoS Negl. Trop. Dis. 2012, 6, e1875. [Google Scholar] [CrossRef]
- Gerardin, P.; Couderc, T.; Bintner, M.; Tournebize, P.; Renouil, M.; Lemant, J.; Boisson, V.; Borgherini, G.; Staikowsky, F.; Schramm, F.; et al. Chikungunya virus-associated encephalitis: A cohort study on La Reunion Island, 2005–2009. Neurology 2016, 86, 94–102. [Google Scholar] [CrossRef]
- Malherbe, H.; Strickland-Cholmley, M.; Jackson, A.L. Sindbis virus infection in man. Report of a case with recovery of virus from skin lesions. S. Afr. Med. J. 1963, 37, 547–552. [Google Scholar]
- Tesh, R.B. Arthritides caused by mosquito-borne viruses. Annu. Rev. Med. 1982, 33, 31–40. [Google Scholar] [CrossRef]
- Gardner, J.; Anraku, I.; Le, T.T.; Larcher, T.; Major, L.; Roques, P.; Schroder, W.A.; Higgs, S.; Suhrbier, A. Chikungunya virus arthritis in adult wild-type mice. J. Virol. 2010, 84, 8021–8032. [Google Scholar] [CrossRef]
- Schilte, C.; Couderc, T.; Chretien, F.; Sourisseau, M.; Gangneux, N.; Guivel-Benhassine, F.; Kraxner, A.; Tschopp, J.; Higgs, S.; Michault, A.; et al. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J. Exp. Med. 2010, 207, 429–442. [Google Scholar] [CrossRef]
- Laurent, P.; Le Roux, K.; Grivard, P.; Bertil, G.; Naze, F.; Picard, M.; Staikowsky, F.; Barau, G.; Schuffenecker, I.; Michault, A. Development of a sensitive real-time reverse transcriptase PCR assay with an internal control to detect and quantify chikungunya virus. Clin. Chem. 2007, 53, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Grivard, P.; Le Roux, K.; Laurent, P.; Fianu, A.; Perrau, J.; Gigan, J.; Hoarau, G.; Grondin, N.; Staikowsky, F.; Favier, F.; et al. Molecular and serological diagnosis of Chikungunya virus infection. Pathol. Biol. 2007, 55, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Touret, Y.; Randrianaivo, H.; Michault, A.; Schuffenecker, I.; Kauffmann, E.; Lenglet, Y.; Barau, G.; Fourmaintraux, A. Early maternal-fetal transmission of the Chikungunya virus. Presse Med. 2006, 35, 1656–1658. [Google Scholar] [CrossRef]
- Pal, P.; Dowd, K.A.; Brien, J.D.; Edeling, M.A.; Gorlatov, S.; Johnson, S.; Lee, I.; Akahata, W.; Nabel, G.J.; Richter, M.K.; et al. Development of a highly protective combination monoclonal antibody therapy against Chikungunya virus. PLoS Pathog. 2013, 9, e1003312. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, A.; Aguilar, P.V.; Bopp, N.E.; Yarovinsky, T.O.; Weaver, S.C.; Rose, J.K. A recombinant virus vaccine that protects against both Chikungunya and Zika virus infections. Vaccine 2018, 36, 3894–3900. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Auguste, A.J.; Kaelber, J.T.; Luo, H.; Rossi, S.L.; Fenton, K.; Leal, G.; Kim, D.Y.; Chiu, W.; Wang, T.; et al. A chikungunya fever vaccine utilizing an insect-specific virus platform. Nat. Med. 2017, 23, 192–199. [Google Scholar] [CrossRef]
- Seymour, R.L.; Rossi, S.L.; Bergren, N.A.; Plante, K.S.; Weaver, S.C. The role of innate versus adaptive immune responses in a mouse model of O’nyong-nyong virus infection. Am. J. Trop. Med. Hyg. 2013, 88, 1170–1179. [Google Scholar] [CrossRef]
- Darwish, M.A.; Hoogstraal, H.; Omar, F.M. A serological survey for Thogoto virus in humans, domestic mammals, and rats in Egypt. J. Egypt. Public Health Assoc. 1979, 54, 1–8. [Google Scholar]
- Filipe, A.R.; Peleteiro, M.C.; Monath, T.M.; Calisher, E.H. Pathological lesions in mice infected with Thogoto virus, a tick-borne orthomyxovirus. Acta Virol. 1986, 30, 337–340. [Google Scholar] [PubMed]
- Buettner, N.; Vogt, C.; Martinez-Sobrido, L.; Weber, F.; Waibler, Z.; Kochs, G. Thogoto virus ML protein is a potent inhibitor of the interferon regulatory factor-7 transcription factor. J. Gen. Virol. 2010, 91, 220–227. [Google Scholar] [CrossRef]
- Hagmaier, K.; Jennings, S.; Buse, J.; Weber, F.; Kochs, G. Novel gene product of Thogoto virus segment 6 codes for an interferon antagonist. J. Virol. 2003, 77, 2747–2752. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Pinilla, E.; Rodriguez-Calvo, T.; Sevilla, N.; Ortego, J. Heterologous prime boost vaccination with DNA and recombinant modified vaccinia virus Ankara protects IFNAR(−/−) mice against lethal bluetongue infection. Vaccine 2009, 28, 437–445. [Google Scholar] [CrossRef]
- Calvo-Pinilla, E.; Navasa, N.; Anguita, J.; Ortego, J. Multiserotype protection elicited by a combinatorial prime-boost vaccination strategy against bluetongue virus. PLoS ONE 2012, 7, e34735. [Google Scholar] [CrossRef] [PubMed]
- Schwartz-Cornil, I.; Mertens, P.P.; Contreras, V.; Hemati, B.; Pascale, F.; Breard, E.; Mellor, P.S.; MacLachlan, N.J.; Zientara, S. Bluetongue virus: Virology, pathogenesis and immunity. Vet. Res. 2008, 39, 46. [Google Scholar] [CrossRef]
- Umeshappa, C.S.; Singh, K.P.; Nanjundappa, R.H.; Pandey, A.B. Apoptosis and immuno-suppression in sheep infected with bluetongue virus serotype-23. Vet. Microbiol. 2010, 144, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Darpel, K.E.; Monaghan, P.; Simpson, J.; Anthony, S.J.; Veronesi, E.; Brooks, H.W.; Elliott, H.; Brownlie, J.; Takamatsu, H.H.; Mellor, P.S.; et al. Involvement of the skin during bluetongue virus infection and replication in the ruminant host. Vet. Res. 2012, 43, 40. [Google Scholar] [CrossRef] [PubMed]
- Worwa, G.; Hilbe, M.; Chaignat, V.; Hofmann, M.A.; Griot, C.; Ehrensperger, F.; Doherr, M.G.; Thur, B. Virological and pathological findings in Bluetongue virus serotype 8 infected sheep. Vet. Microbiol. 2010, 144, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Howerth, E.W.; Tyler, D.E. Experimentally induced bluetongue virus infection in white-tailed deer: Ultrastructural findings. Am. J. Vet. Res. 1988, 49, 1914–1922. [Google Scholar]
- Caporale, M.; Wash, R.; Pini, A.; Savini, G.; Franchi, P.; Golder, M.; Patterson-Kane, J.; Mertens, P.; Di Gialleonardo, L.; Armillotta, G.; et al. Determinants of bluetongue virus virulence in murine models of disease. J. Virol. 2011, 85, 11479–11489. [Google Scholar] [CrossRef] [PubMed]
- Caporale, M.; Di Gialleonorado, L.; Janowicz, A.; Wilkie, G.; Shaw, A.; Savini, G.; Van Rijn, P.A.; Mertens, P.; Di Ventura, M.; Palmarini, M. Virus and host factors affecting the clinical outcome of bluetongue virus infection. J. Virol. 2014, 88, 10399–10411. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, V.; Capocefalo, A.; Calvo-Pinilla, E.; Redaelli, M.; Mucignat-Caretta, C.; Mertens, P.; Ortego, J.; Donofrio, G. Immunization of knock-out alpha/beta interferon receptor mice against lethal bluetongue infection with a BoHV-4-based vector expressing BTV-8 VP2 antigen. Vaccine 2011, 29, 3074–3082. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Eschbaumer, M.; Said, A.; Hoffmann, B.; Beer, M.; Osterrieder, N. An equine herpesvirus type 1 (EHV-1) expressing VP2 and VP5 of serotype 8 bluetongue virus (BTV-8) induces protection in a murine infection model. PLoS ONE 2012, 7, e34425. [Google Scholar] [CrossRef] [PubMed]
- Jabbar, T.K.; Calvo-Pinilla, E.; Mateos, F.; Gubbins, S.; Bin-Tarif, A.; Bachanek-Bankowska, K.; Alpar, O.; Ortego, J.; Takamatsu, H.H.; Mertens, P.P.; et al. Protection of IFNAR(−/−) mice against bluetongue virus serotype 8, by heterologous (DNA/rMVA) and homologous (rMVA/rMVA) vaccination, expressing outer-capsid protein VP2. PLoS ONE 2013, 8, e60574. [Google Scholar] [CrossRef]
- Marin-Lopez, A.; Otero-Romero, I.; de la Poza, F.; Menaya-Vargas, R.; Calvo-Pinilla, E.; Benavente, J.; Martinez-Costas, J.M.; Ortego, J. VP2, VP7, and NS1 proteins of bluetongue virus targeted in avian reovirus muNS-Mi microspheres elicit a protective immune response in IFNAR(−/−) mice. Antivir. Res. 2014, 110, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Mohd Jaafar, F.; Belhouchet, M.; Vitour, D.; Adam, M.; Breard, E.; Zientara, S.; Mertens, P.P.; Attoui, H. Immunisation with bacterial expressed VP2 and VP5 of bluetongue virus (BTV) protect alpha/beta interferon-receptor knock-out (IFNAR(−/−)) mice from homologous lethal challenge. Vaccine 2014, 32, 4059–4067. [Google Scholar] [CrossRef]
- Martin, V.; Pascual, E.; Avia, M.; Pena, L.; Valcarcel, F.; Sevilla, N. Protective Efficacy in Sheep of Adenovirus-Vectored Vaccines against Bluetongue Virus Is Associated with Specific T Cell Responses. PLoS ONE 2015, 10, e0143273. [Google Scholar] [CrossRef]
- Marin-Lopez, A.; Calvo-Pinilla, E.; Barriales, D.; Lorenzo, G.; Benavente, J.; Brun, A.; Martinez-Costas, J.M.; Ortego, J. Microspheres-prime/rMVA-boost vaccination enhances humoral and cellular immune response in IFNAR(−/−) mice conferring protection against serotypes 1 and 4 of bluetongue virus. Antivir. Res. 2017, 142, 55–62. [Google Scholar] [CrossRef]
- Marin-Lopez, A.; Calvo-Pinilla, E.; Barriales, D.; Lorenzo, G.; Brun, A.; Anguita, J.; Ortego, J. CD8 T Cell Responses to an Immunodominant Epitope within the Nonstructural Protein NS1 Provide Wide Immunoprotection against Bluetongue Virus in IFNAR(−/−) Mice. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Li, J.; Yang, T.; Xu, Q.; Sun, E.; Feng, Y.; Lv, S.; Zhang, Q.; Wang, H.; Wu, D. DNA vaccine prime and recombinant FPV vaccine boost: An important candidate immunization strategy to control bluetongue virus type 1. Appl. Microbiol. Biotechnol. 2015, 99, 8643–8652. [Google Scholar] [CrossRef]
- Legisa, D.M.; Perez Aguirreburualde, M.S.; Gonzalez, F.N.; Marin-Lopez, A.; Ruiz, V.; Wigdorovitz, A.; Martinez-Escribano, J.A.; Ortego, J.; Dus Santos, M.J. An experimental subunit vaccine based on Bluetongue virus 4 VP2 protein fused to an antigen-presenting cells single chain antibody elicits cellular and humoral immune responses in cattle, guinea pigs and IFNAR(−/−) mice. Vaccine 2015, 33, 2614–2619. [Google Scholar] [CrossRef]
- Van Zyl, A.R.; Meyers, A.E.; Rybicki, E.P. Development of plant-produced protein body vaccine candidates for bluetongue virus. BMC Biotechnol. 2017, 17, 47. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, D.K.A.; Du, J.; Gao, S.; Tian, Z.; Zhang, G.; Huang, D.; Du, R.; Kang, B.; Liu, G.; Luo, J.; et al. Evaluation of the immune response afforded by a subunit vaccine candidate against bluetongue virus in mice and sheep. Vet. Microbiol. 2018, 219, 40–48. [Google Scholar] [CrossRef] [PubMed]
- House, J.A. African horse sickness. Vet. Clin. N. Am. Equine Pract. 1993, 9, 355–364. [Google Scholar] [CrossRef]
- O’Hara, R.S.; Meyer, A.J.; Burroughs, J.N.; Pullen, L.; Martin, L.A.; Mertens, P.P. Development of a mouse model system, coding assignments and identification of the genome segments controlling virulence of African horse sickness virus serotypes 3 and 8. Arch. Virol. Suppl. 1998, 14, 259–279. [Google Scholar] [PubMed]
- De la Grandiere, M.A.; Dal Pozzo, F.; Tignon, M.; Zonta, W.; Thiry, D.; Mauroy, A.; Mathijs, E.; Caij, A.B.; Saegerman, C.; Thiry, E. Study of the virulence of serotypes 4 and 9 of African horse sickness virus in IFNAR(−/−), Balb/C and 129 Sv/Ev mice. Vet. Microbiol. 2014, 174, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P.; Cropp, C.B.; Harrison, A.K. Mode of entry of a neurotropic arbovirus into the central nervous system. Reinvestigation of an old controversy. Lab. Investig. 1983, 48, 399–410. [Google Scholar] [PubMed]
- Mellor, P.S.; Hamblin, C. African horse sickness. Vet. Res. 2004, 35, 445–466. [Google Scholar] [CrossRef] [PubMed]
- Lulla, V.; Lulla, A.; Wernike, K.; Aebischer, A.; Beer, M.; Roy, P. Assembly of Replication-Incompetent African Horse Sickness Virus Particles: Rational Design of Vaccines for All Serotypes. J. Virol. 2016, 90, 7405–7414. [Google Scholar] [CrossRef] [PubMed]
- Alberca, B.; Bachanek-Bankowska, K.; Cabana, M.; Calvo-Pinilla, E.; Viaplana, E.; Frost, L.; Gubbins, S.; Urniza, A.; Mertens, P.; Castillo-Olivares, J. Vaccination of horses with a recombinant modified vaccinia Ankara virus (MVA) expressing African horse sickness (AHS) virus major capsid protein VP2 provides complete clinical protection against challenge. Vaccine 2014, 32, 3670–3674. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Pinilla, E.; de la Poza, F.; Gubbins, S.; Mertens, P.P.; Ortego, J.; Castillo-Olivares, J. Vaccination of mice with a modified Vaccinia Ankara (MVA) virus expressing the African horse sickness virus (AHSV) capsid protein VP2 induces virus neutralising antibodies that confer protection against AHSV upon passive immunisation. Virus Res. 2014, 180, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Pinilla, E.; de la Poza, F.; Gubbins, S.; Mertens, P.P.; Ortego, J.; Castillo-Olivares, J. Antiserum from mice vaccinated with modified vaccinia Ankara virus expressing African horse sickness virus (AHSV) VP2 provides protection when it is administered 48 h before, or 48 h after challenge. Antivir. Res. 2015, 116, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Shope, R.E.; Macnamara, L.G.; Mangold, R. A Virus-Induced Epizootic Hemorrhagic Disease of the Virginia White-Tailed Deer (Odocoileus Virginianus). J. Exp. Med. 1960, 111, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Mettler, N.E.; Macnamara, L.G.; Shope, R.E. The propagation of the virus of epizootic hemorrhagic disease of deer in newborn mice and HeLa cells. J. Exp. Med. 1962, 116, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, A.M.; Randolph, S.E. Drivers, dynamics, and control of emerging vector-borne zoonotic diseases. Lancet 2012, 380, 1946–1955. [Google Scholar] [CrossRef]
- Kolumam, G.A.; Thomas, S.; Thompson, L.J.; Sprent, J.; Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 2005, 202, 637–650. [Google Scholar] [CrossRef]
- Tough, D.F.; Borrow, P.; Sprent, J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science 1996, 272, 1947–1950. [Google Scholar] [CrossRef]
- Wang, Y.; Swiecki, M.; Cella, M.; Alber, G.; Schreiber, R.D.; Gilfillan, S.; Colonna, M. Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe 2012, 11, 631–642. [Google Scholar] [CrossRef]


{kind=link}
| 9 | Serotype or Strain | Mortality | Clinical Signs | Vaccine Model |
|---|---|---|---|---|
| Rift Valley fever virus [29,30,31,32,33] | ZH548, MP12, Clone 13 | Yes | Swollen and congested liver, acute hepatitis. Ruffled fur, hunched posture, and lethargy | DNA-Gn/Gc |
| Crimean Congo Fever Virus [5,34,35,36,37,38,39,40] | IbAr 2000, IbAr 10200 | Yes | Labored breathing and porphyry around the nostrils and eyes. Organ pathology (liver and lymphoid tissue), thrombocytopenia, coagulopathy, weight loss, ruffled fur, hunched posture, and lethargy. | CCHFV alum-adjuvanted vaccines, VLPs, DNA or viral vector vaccines (MVA and adenovirus) expressing nucleocapsid protein or glycoproteins |
| Schmallenberg virus [41,42,43] | wild-type SBV (wtSBV), isolate BH80/11 | Partial (50%) | Weight loss, ataxia, and apathy. | DNA-Gn/Gc/N, DNA-N-terminal GC, recombinant-N-terminal GC |
| Dengue virus [44,45,46,47,48] | DENV-1 DENV-2 DENV-3 DENV-4 | Yes Yes ND No | Severe dengue-like disease. | Live attenuated mutants in the 2′-O-methyltransferase (2′-O-MTase) of DENV-1 and DENV-2 |
| Yellow fever virus [49] | Asibi or Angola73 | Yes | Viscerotropic disease. | ND |
| Zika virus [50,51,52,53,54,55] | MP1751 H/PF/2013 MR 766. 5 ZIKV-Paraiba | Yes | Severe disease, including hind limb weakness and paralysis. | Vaccinia-based single vector encoding polyprotein DNA-prME |
| West Nile virus [28,56] | WNV strain 3000.0259 | Yes | Hunched posture, ruffled fur and reduced activities. Encephalitis. | RepliVAX WN, single-cycle West Nile vaccine |
| Japanese encephalitis virus [57] | JaOArS982 | Yes | Slow movement, ataxia, piloerection, anorexia and continuous weight loss. | ND |
| Chikungunya virus [58] | CHIKV-21 | Yes | Weakness of the limbs (loss of muscle tone) and lethargy. | VSV-CHIKV-E3-E2-6K-E1 EILV/CHIKV chimeras |
| Sindbis virus [59] | TR339 | Yes | Weight loss and fur ruffling. | ND |
| Venezuelan equine encephalitis [60] | V-3000 | Yes | Pronounced hunching, lethargy, prostration, and death. | ND |
| Vesicular stomatitis virus [61,62] | VSV Indiana | Yes | Neuropathy. | ND |
| Thogoto virus [63,64] | Yes | Pathological lesions in the lungs, liver and intestine. | ND | |
| Bluetongue virus [6,65,66] | BTV-1 BTV-2 BTV-4 BTV-8 BTV-16 | Yes | Splenomegaly, congested lung. Hunched posture, ruffled fur, conjunctivitis. | DNA, Herpesvirus Poxvirus, Baculovirus, and bacterial expressed proteins, Adenovirus |
| African horse sickness virus [67,68] | AHSV-1 AHSV-3 AHSV-4 AHSV-9 | Yes | Ruffled fur, lethargy, ocular discharges, hemorrhages in lung, splenomegaly, congestion of liver. | DNA, Poxvirus |
| Epizootic hemorrhagic disease virus [69] | EHDV-7 | Yes | Splenomegaly, necrotic foci in the liver. | ND |
| Vaccine Based on | Protein Expressed | Protection against Homologous BTV | Protection against Heterologous BTV | Reference |
|---|---|---|---|---|
| BTV inactivated vaccine | - | Yes | Not determined | Calvo-Pinilla et al., 2009 [6] |
| MVA virus | VP2 and VP5 | Partial | No | Calvo-Pinilla, 2009 [134] |
| Bovine herpes virus | VP2 | Partial | No | Franceschi et al., 2011 [143] |
| Equine herpes virus | VP2 and VP5 | Partial | No | Ma et al., 2012 [144] |
| MVA virus | VP2, VP5, and VP7 | Yes | No | Calvo-Pinilla et al., 2009 [134] Jabbar et al., 2013 [145] |
| MVA virus | VP2, VP7, and NS1 | Yes | Yes | Calvo-Pinilla et al., 2012 [135] |
| muNs microspheres | VP2, VP7, and NS1 | Yes | Partial | Marín-López et al., 2014 [146] |
| Bacterial expressed proteins | VP2 domains | Yes | No | Mohd Jaafar et al., 2014 [147] |
| Adenovirus | VP2, VP7, and NS3 | Yes | ND | Martín et al., 2015 [148] |
| muNS/MVA virus | VP2, VP7, and NS1 | Yes | Yes | Marín-López et al., 2017 [149] |
| MVA virus | NS1 | Yes | Yes | Marín-López et al., 2018 [150] |
| DNA/Fowlpox virus | VP2 and VP5 | ND | ND | Li et al., 2015 [151] |
| Baculovirus expressed proteins | VP2 alone or fused to APCH | ND | ND | Legisa et al., 2015 [152] |
| Plant-produced protein | VP2 alone or VP2 B-cell epitope sequences | ND | ND | van Zyl et al., 2017 [153] |
| Bacterial and baculovirus expressed proteins | VP2, VP3, VP7, NS2, truncated VP5 | ND | ND | Mohamed et al., 2018 [154] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marín-Lopez, A.; Calvo-Pinilla, E.; Moreno, S.; Utrilla-Trigo, S.; Nogales, A.; Brun, A.; Fikrig, E.; Ortego, J. Modeling Arboviral Infection in Mice Lacking the Interferon Alpha/Beta Receptor. Viruses 2019, 11, 35. https://doi.org/10.3390/v11010035
Marín-Lopez A, Calvo-Pinilla E, Moreno S, Utrilla-Trigo S, Nogales A, Brun A, Fikrig E, Ortego J. Modeling Arboviral Infection in Mice Lacking the Interferon Alpha/Beta Receptor. Viruses. 2019; 11(1):35. https://doi.org/10.3390/v11010035
Chicago/Turabian StyleMarín-Lopez, Alejandro, Eva Calvo-Pinilla, Sandra Moreno, Sergio Utrilla-Trigo, Aitor Nogales, Alejandro Brun, Erol Fikrig, and Javier Ortego. 2019. "Modeling Arboviral Infection in Mice Lacking the Interferon Alpha/Beta Receptor" Viruses 11, no. 1: 35. https://doi.org/10.3390/v11010035
APA StyleMarín-Lopez, A., Calvo-Pinilla, E., Moreno, S., Utrilla-Trigo, S., Nogales, A., Brun, A., Fikrig, E., & Ortego, J. (2019). Modeling Arboviral Infection in Mice Lacking the Interferon Alpha/Beta Receptor. Viruses, 11(1), 35. https://doi.org/10.3390/v11010035