US28: HCMV’s Swiss Army Knife


Abstract
1. Chemokine Receptors
2. Expression of HCMV-Encoded Chemokine Receptor Homologues and Their Interactions
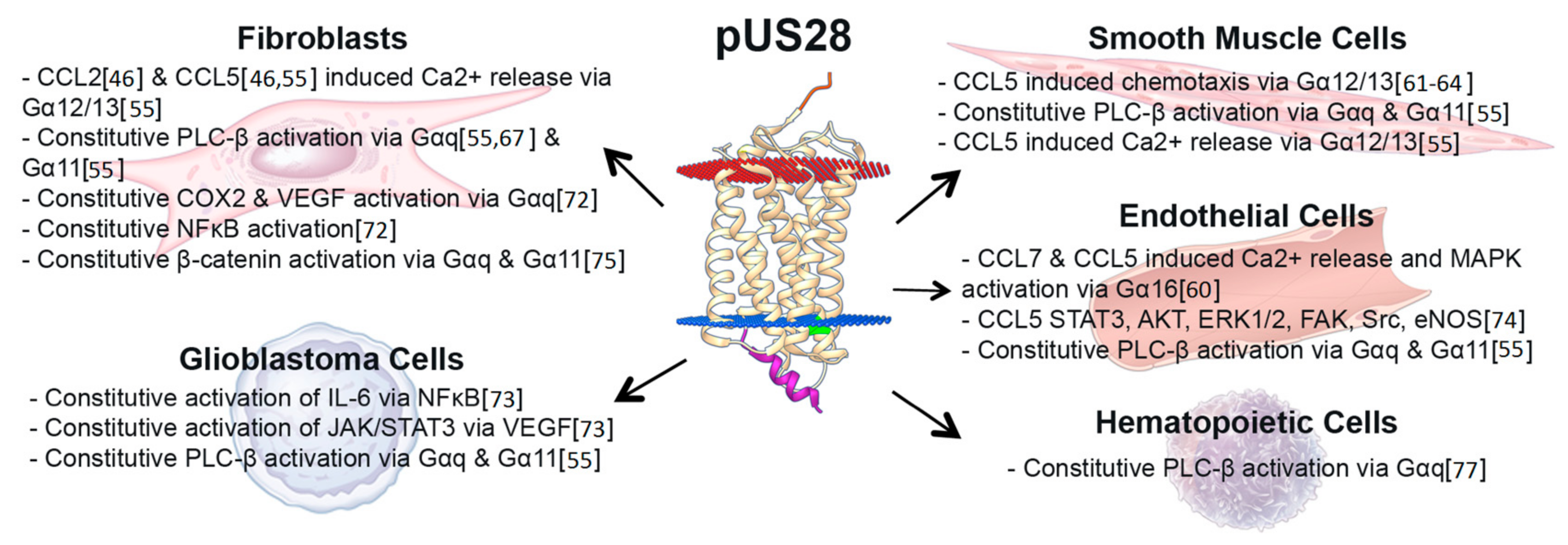
3. US28 Constitutive Signaling during Lytic Infection Activates Multiple Distinct Signaling Pathways, Altering the Infected Host Cell Environment
4. Chemokine Ligand-Induced pUS28-Mediated Signaling during Lytic Infection
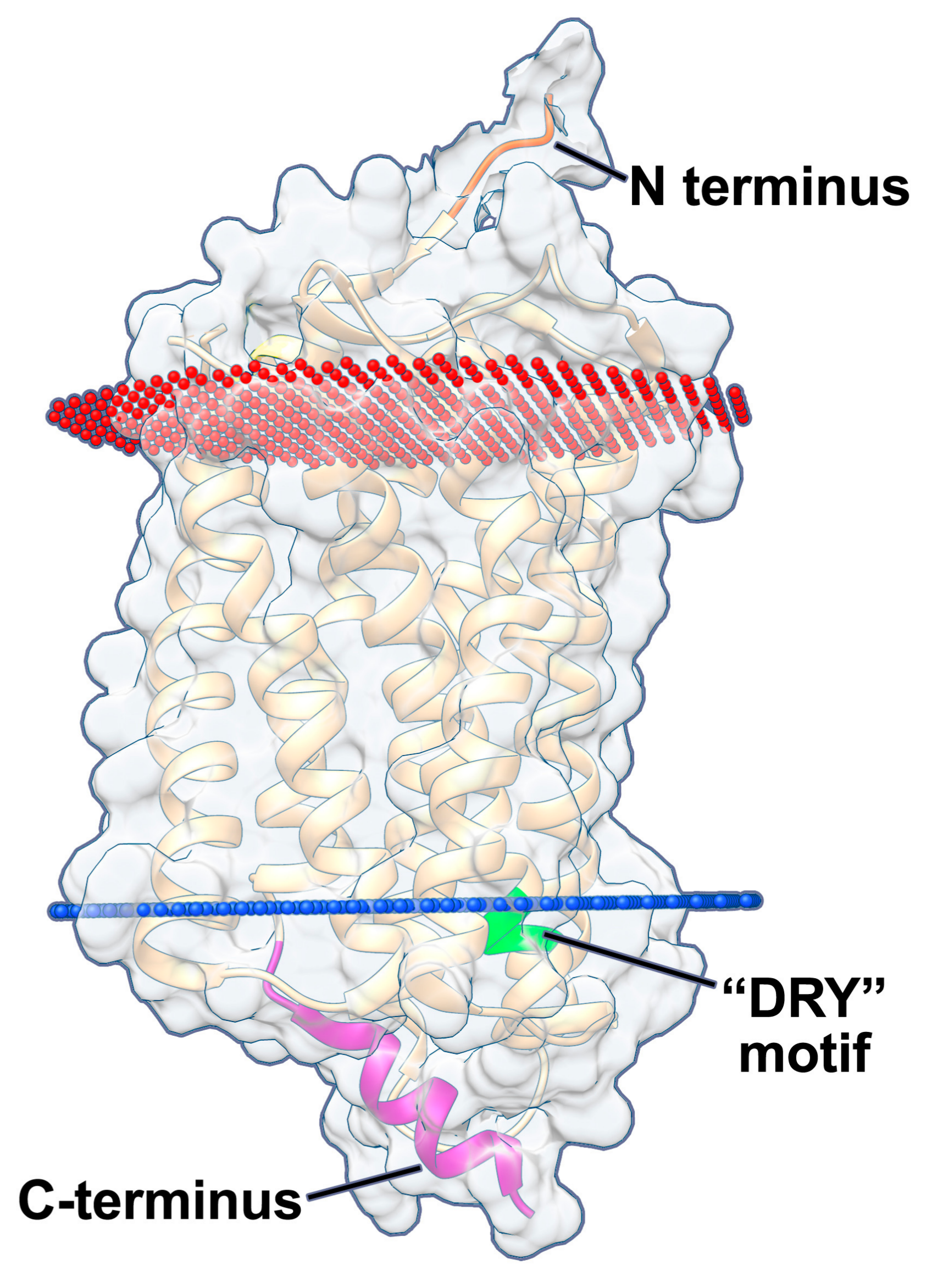
5. US28 Localization during Lytic Infection
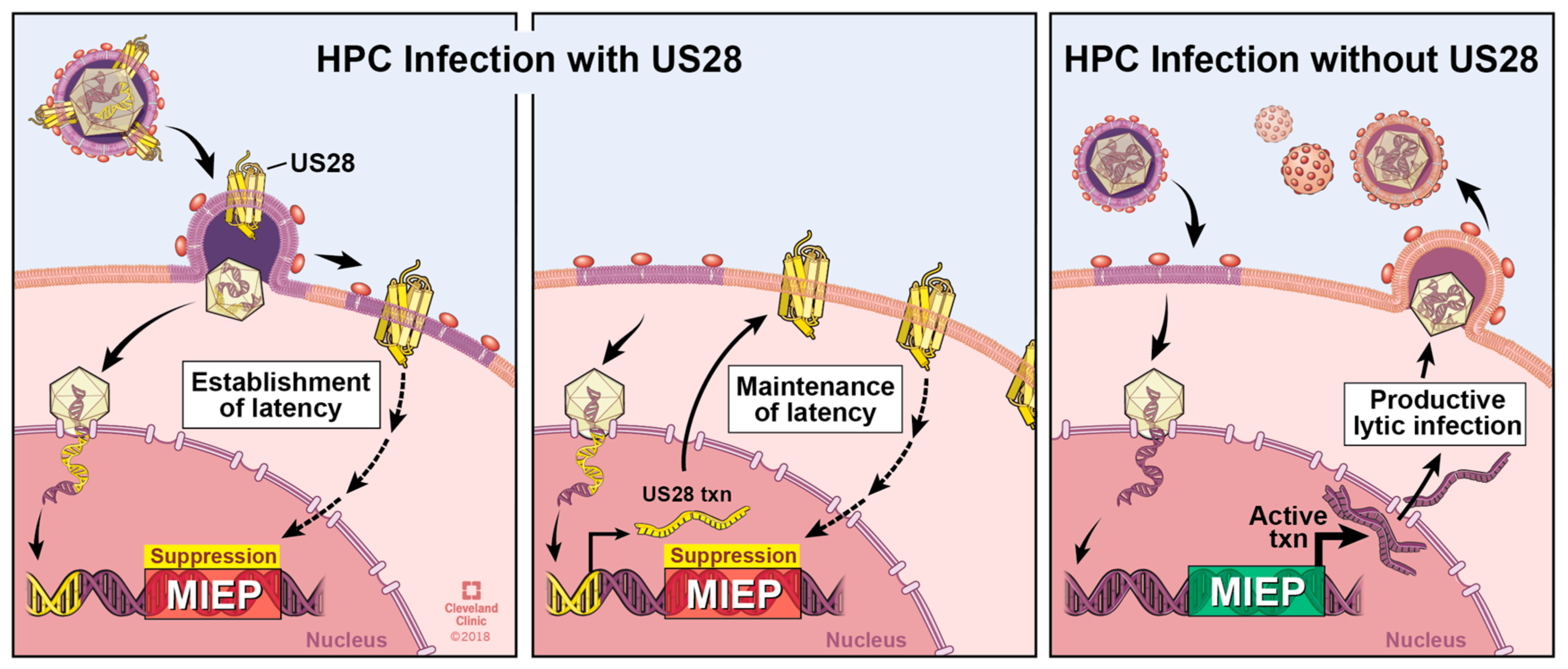
6. The Roles of pUS28 in Latent Infection
7. US28 and Cancers
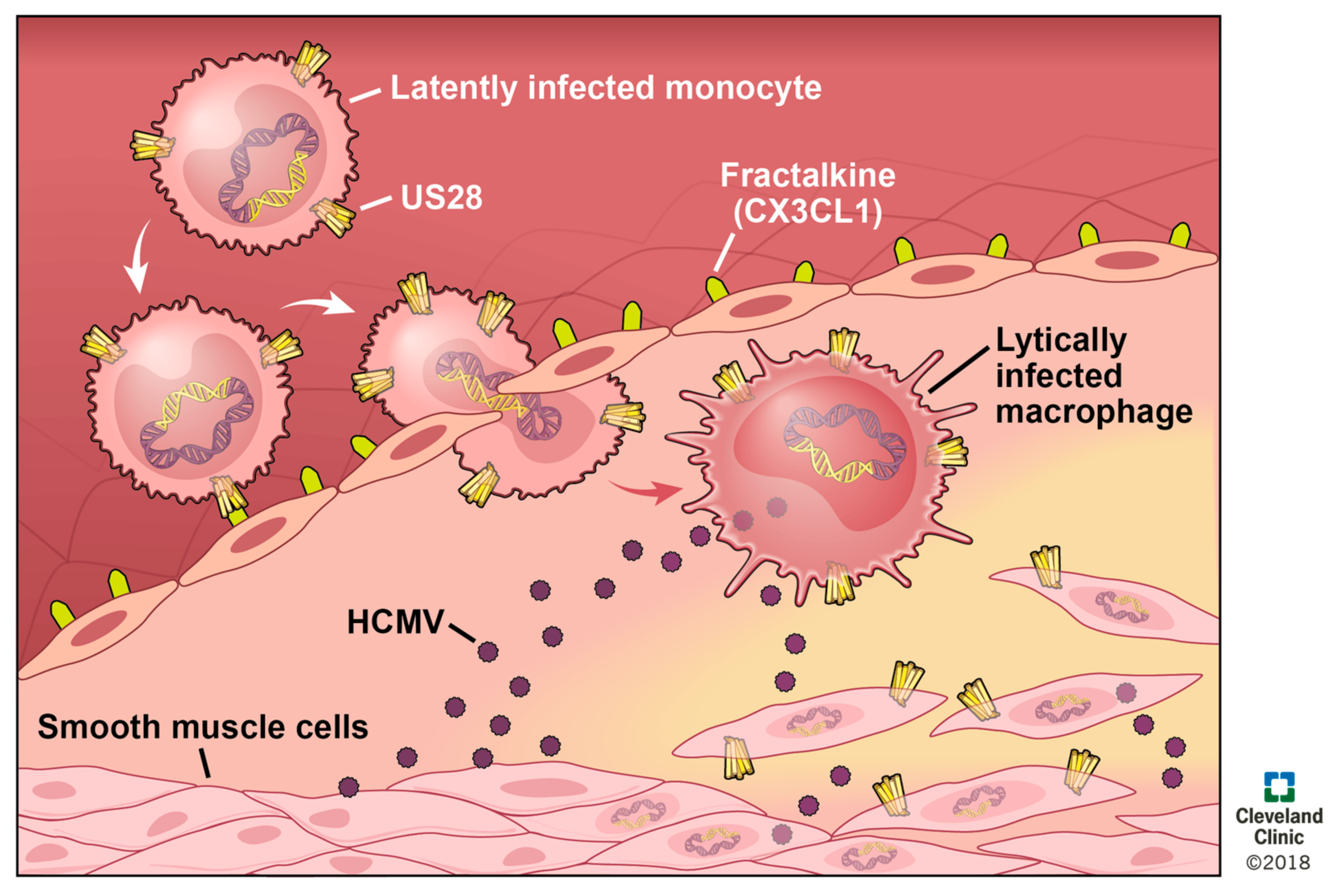
8. US28 in Vascular Disease
9. US28 as a Therapeutic Target
10. Concluding Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Melnick, J.L.; Adam, E.; DeBakey, M.E. Cytomegalovirus and atherosclerosis. Arch. Immunol. Ther. Exp. 1996, 44, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Doerr, H.W.; Cinatl, J. Oncomodulation by human cytomegalovirus: Evidence becomes stronger. Med. Microbiol. Immunol. 2009, 198, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Mestas, E. Congenital cytomegalovirus. Adv. Neonatal Care 2016, 16, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Marty, F.M.; Ljungman, P.; Chemaly, R.F.; Maertens, J.; Dadwal, S.S.; Duarte, R.F.; Haider, S.; Ullmann, A.J.; Katayama, Y.; Brown, J. Letermovir prophylaxis for cytomegalovirus in hematopoietic-cell transplantation. N. Engl. J. Med. 2017, 377, 2433–2444. [Google Scholar] [CrossRef] [PubMed]
- Lischka, P.; Zimmermann, H. Antiviral strategies to combat cytomegalovirus infections in transplant recipients. Curr. Opin. Pharmacol. 2008, 8, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Humar, A.; Lebranchu, Y.; Vincenti, F.; Blumberg, E.A.; Punch, J.D.; Limaye, A.P.; Abramowicz, D.; Jardine, A.G.; Voulgari, A.T.; Ives, J.; et al. The efficacy and safety of 200 days valganciclovir cytomegalovirus prophylaxis in high-risk kidney transplant recipients. Am. J. Transplant. 2010, 10, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Alcami, A. Viral mimicry of cytokines, chemokines and their receptors. Nat. Rev. Immunol. 2003, 3, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Chee, M.S.; Bankier, A.T.; Beck, S.; Bohni, R.; Brown, C.M.; Cerny, R.; Horsnell, T.; Hutchison, C.A., 3rd; Kouzarides, T.; Martignetti, J.A. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol. 1990, 154, 125–169. [Google Scholar] [PubMed]
- Gompels, U.A.; Nicholas, J.; Lawrence, G.; Jones, M.; Thomson, B.J.; Martin, M.E.D.; Efstathiou, S.; Craxton, M.; Macaulay, H.A. The DNA sequence of human herpesvirus-6: Structure, coding content, and genome evolution. Virology 1995, 209, 29–51. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Bray, D.; Hopkin, K.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Cell communication. In Essential Cell Biology, 3rd ed.; Garland Science: New York, NY, USA, 2013; pp. 539–549. [Google Scholar]
- Zhang, X.; Eggert, U.S. Non-traditional roles of G protein-coupled receptors in basic cell biology. Mol. Biosyst. 2013, 9, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Drews, J.U. Drug discovery: A historical perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Tautermann, C.S. GPCR structures in drug design, emerging opportunities with new structures. Bioorg. Med. Chem. Lett. 2014, 24, 4073–4079. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR drug targets. Cell 2018, 172, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M. Chemokines and leukocyte traffic. Nature 1998, 392, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.M. International union of pharmacology. XXX. Update on chemokine receptor nomenclature. Pharmacol. Rev. 2002, 54, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Colobran, R.; Pujol-Borrell, R.; Armengol, M.P.; Juan, M. The chemokine network. I. How the genomic organization of chemokines contains clues for deciphering their functional complexity. Clin. Exp. Immunol. 2007, 148, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.M.; Baggiolini, M.; Charo, I.F.; Hébert, C.A.; Horuk, R.; Matsushima, K.; Miller, L.H.; Oppenheim, J.J.; Power, C.A. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev. 2000, 52, 145–176. [Google Scholar] [PubMed]
- Ahuja, S.K.; Lee, J.C.; Murphy, P.M. CXC chemokines bind to unique sets of selectivity determinants that can function independently and are broadly distributed on multiple domains of human interleukin-8 receptor b determinants of high affinity binding and receptor activation are distinct. J. Biol. Chem. 1996, 271, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Monteclaro, F.S.; Charo, I.F. The amino-terminal extracellular domain of the MCP-1 receptor, but not the RANTES/MIP-1α receptor, confers chemokine selectivity evidence for a two-step mechanism for MCP-1 receptor activation. J. Biol. Chem. 1996, 271, 19084–19092. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.K.; Allen, S.; Hsu, A.R.; Handel, T.M. Chemokine-receptor interactions: GPCRs, glycosaminoglycans and viral chemokine binding proteins. Adv. Protein Chem. 2004, 68, 351–391. [Google Scholar] [PubMed]
- Saidak, Z.; Blake-Palmer, K.; Hay, D.L.; Northup, J.K.; Glass, M. Differential activation of G-proteins by μ-opioid receptor agonists. Br. J. Pharmacol. 2006, 147, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Woo, A.Y.-H.; Wang, T.-B.; Zeng, X.; Zhu, W.; Abernethy, D.R.; Wainer, I.W.; Xiao, R.-P. Stereochemistry of an agonist determines coupling preference of β2-adrenoceptor to different G proteins in cardiomyocytes. Mol. Pharmacol. 2009, 75, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Brass, L.F.; Manning, D.R. The gq and g12 families of heterotrimeric G proteins report functional selectivity. Mol. Pharmacol. 2009, 75, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Auger, G.A.; Pease, J.E.; Shen, X.; Xanthou, G.; Barker, M.D. Alanine scanning mutagenesis of CCR3 reveals that the three intracellular loops are essential for functional receptor expression. Eur. J. Immunol. 2002, 32, 1052–1058. [Google Scholar] [CrossRef]
- Haskell, C.A.; Cleary, M.D.; Charo, I.F. Molecular uncoupling of fractalkine-mediated cell adhesion and signal transduction rapid flow arrest of CX3CR1-expressing cells is independent of G-protein activation. J. Biol. Chem. 1999, 274, 10053–10058. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, T.W.; Frimurer, T.M.; Holst, B.; Rosenkilde, M.M.; Elling, C.E. Molecular mechanism of 7TM receptor activation—A global toggle switch model. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 481–519. [Google Scholar] [CrossRef] [PubMed]
- Rovati, G.E.; Capra, V.; Neubig, R.R. The highly conserved dry motif of class a G protein-coupled receptors: Beyond the ground state. Mol. Pharmacol. 2007, 71, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Tobin, A.B.; Butcher, A.J.; Kong, K.C. Location, location, location…site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends. Pharmacol. Sci. 2008, 29, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Vomaske, J.; Nelson, J.A.; Streblow, D.N. Human cytomegalovirus US28: A functionally selective chemokine binding receptor. Infect. Disord. Drug Targets 2009, 9, 548. [Google Scholar] [CrossRef] [PubMed]
- Krupnick, J.G.; Benovic, J.L. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 289–319. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, J.A.; Freedman, N.J.; Lefkowitz, R.J. G protein-coupled receptor kinases. Annu. Rev. Biochem. 1998, 67, 653–692. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Bailey, C.P.; Henderson, G. Agonist-selective mechanisms of GPCR desensitization. Br. J. Pharmacol. 2008, 153, S379–S388. [Google Scholar] [CrossRef] [PubMed]
- Burg, J.S.; Ingram, J.R.; Venkatakrishnan, A.J.; Jude, K.M.; Dukkipati, A.; Feinberg, E.N.; Angelini, A.; Waghray, D.; Dror, R.O.; Ploegh, H.L.; et al. Structural basis for chemokine recognition and activation of a viral G protein—Coupled receptor. Science 2015, 347, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Preininger, A.M.; Hamm, H.E. G protein signaling: Insights from new structures. Sci. STKE 2004, 2014, re3. [Google Scholar] [CrossRef] [PubMed]
- Timothy, A.; Casey, P.J. Signalling functions and biochemical properties of pertussis toxin-resistant G-proteins. Biochem. J. 1997, 321, 561–571. [Google Scholar]
- Welch, A.R.; McGregor, L.M.; Gibson, W. Cytomegalovirus homologs of cellular G protein-coupled receptor genes are transcribed. J. Virol. 1991, 65, 3915–3918. [Google Scholar] [PubMed]
- Margulies, B.J.; Browne, H.; Gibson, W. Identification of the human cytomegalovirus G protein-coupled receptor homologue encoded by UL33 in infected cells and enveloped virus particles. Virology 1996, 225, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Tadagaki, K.; Tudor, D.; Gbahou, F.; Tschische, P.; Waldhoer, M.; Bomsel, M.; Jockers, R.; Kamal, M. Human cytomegalovirus-encoded UL33 and UL78 heteromerize with host CCR5 and CXCR4 impairing their HIV coreceptor activity. Blood 2012, 119, 4908–4918. [Google Scholar] [CrossRef] [PubMed]
- Arnolds, K.L.; Lares, A.P.; Spencer, J.V. The US27 gene product of human cytomegalovirus enhances signaling of host chemokine receptor CXCR4. Virology 2013, 439, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef] [PubMed]
- Michel, D.; Milotić, I.; Wagner, M.; Vaida, B.; Holl, J.; Ansorge, R.; Mertens, T. The human cytomegalovirus UL78 gene is highly conserved among clinical isolates, but is dispensable for replication in fibroblasts and a renal artery organ-culture system. J. Gen. Virol. 2005, 86, 297–306. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Shenk, T. Human cytomegalovirus PUL78 G protein-coupled receptor homologue is required for timely cell entry in epithelial cells but not fibroblasts. J. Virol. 2012, 86, 11425–11433. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.; Schall, T.J.; Corey, L.; Geballe, A.P. Functional analysis of the human cytomegalovirus US28 gene by insertion mutagenesis with the green fluorescent protein gene. J. Virol. 1998, 72, 8158–8165. [Google Scholar] [PubMed]
- O’Connor, C.M.; Shenk, T. Human cytomegalovirus PUS27 G protein-coupled receptor homologue is required for efficient spread by the extracellular route but not for direct cell-to-cell spread. J. Virol. 2011, 85, 3700–3707. [Google Scholar] [CrossRef] [PubMed]
- Noriega, V.M.; Gardner, T.J.; Redmann, V.; Bongers, G.; Lira, S.A.; Tortorella, D. Human cytomegalovirus US28 facilitates cell-to-cell viral dissemination. Viruses 2014, 6, 1202–1218. [Google Scholar] [CrossRef] [PubMed]
- Waldhoer, M.; Kledal, T.N.; Farrell, H.; Schwartz, T.W. Murine cytomegalovirus (CMV) M33 and human CMV US28 receptors exhibit similar constitutive signaling activities. J. Virol. 2002, 76, 8161–8168. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.C.; Arnolds, K.L.; O’Connor, C.M.; Spencer, J.V. Human cytomegalovirus UL111A and US27 gene products enhance the CXCL12/CXCR4 signaling axis via distinct mechanisms. J. Virol. 2018, 92, e01981–e01917. [Google Scholar] [CrossRef] [PubMed]
- Casarosa, P.; Menge, W.M.; Minisini, R.; Otto, C.; van Heteren, J.; Jongejan, A.; Timmerman, H.; Moepps, B.; Kirchhoff, F.; Mertens, T.; et al. Identification of the first nonpeptidergic inverse agonist for a constitutively active viral-encoded G protein-coupled receptor. J. Biol. Chem. 2003, 278, 5172–5178. [Google Scholar] [CrossRef] [PubMed]
- Humby, M.S.; O’Connor, C.M. HCMV US28 is important for latent infection of hemtopoietic progenitor cells. J. Virol. 2015. [Google Scholar] [CrossRef]
- Cheng, S.; Caviness, K.; Buehler, J.; Smithey, M.; Nikolich-Zugich, J.; Goodrum, F. Transcriptome-wide characterization of human cytomegalovirus in natural infection and experimental latency. Proc. Natl. Acad. Sci. USA 2017, 114, E10586–E10595. [Google Scholar] [CrossRef] [PubMed]
- Tschische, P.; Tadagaki, K.; Kamal, M.; Jockers, R.; Waldhoer, M. Heteromerization of human cytomegalovirus encoded chemokine receptors. Biochem. Pharmacol. 2011, 82, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.E.; Zagorski, W.A.; Brenneman, J.D.; Avery, D.; Miller, J.L.C.; O’Connor, C.M. US28 is a potent activator of phospholipase C during HCMV infection of clinically relevant target cells. PLoS ONE 2012, 7, e50524. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Poole, E.L.; Jackson, S.E.; Smit, M.J.; Wills, M.R.; Sinclair, J.H. Latency-associated expression of human cytomegalovirus US28 attenuates cell signaling pathways to maintain latent infection. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G. G protein-coupled receptor dimerization: Function and ligand pharmacology. Mol. Pharmacol. 2004, 66, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Prinster, S.C.; Hague, C.; Hall, R.A. Heterodimerization of G protein-coupled receptors: Specificity and functional significance. Pharmacol. Rev. 2005, 57, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.-L.; Murphy, P.M. Human cytomegalovirus open reading frame US28 encodes a functional beta chemokine receptor. J. Biol. Chem. 1994, 269, 28539–28542. [Google Scholar] [PubMed]
- Billstrom, M.A.; Johnson, G.L.; Avdi, N.J.; Worthen, G.S. Intracellular signaling by the chemokine receptor US28 during human cytomegalovirus infection. J. Virol. 1998, 72, 5535–5544. [Google Scholar] [PubMed]
- Streblow, D.N.; Soderberg-Naucler, C.; Vieira, J.; Smith, P.; Wakabayashi, E.; Ruchti, F.; Mattison, K.; Altschuler, Y.; Nelson, J.A. The human cytomegalovirus chemokine receptor US28 mediates vascular smooth muscle cell migration. Cell 1999, 99, 511–520. [Google Scholar] [CrossRef]
- Streblow, D.N.; Vomaske, J.; Smith, P.; Melnychuk, R.; Hall, L.; Pancheva, D.; Smit, M.; Casarosa, P.; Schlaepfer, D.D.; Nelson, J.A. Human cytomegalovirus chemokine receptor US28-induced smooth muscle cell migration is mediated by focal adhesion kinase and SRC. J. Biol. Chem. 2003, 278, 50456–50465. [Google Scholar] [CrossRef] [PubMed]
- Melnychuk, R.M.; Streblow, D.N.; Smith, P.P.; Hirsch, A.J.; Pancheva, D.; Nelson, J.A. Human cytomegalovirus-encoded G protein-coupled receptor US28 mediates smooth muscle cell migration through Gα12. J. Virol. 2004, 78, 8382–8391. [Google Scholar] [CrossRef] [PubMed]
- Vomaske, J.; Melnychuk, R.M.; Smith, P.P.; Powell, J.; Hall, L.; DeFilippis, V.; Fruh, K.; Smit, M.; Schlaepfer, D.D.; Nelson, J.A.; et al. Differential ligand binding to a human cytomegalovirus chemokine receptor determines cell type-specific motility. PLoS Pathog. 2009, 5, e1000304. [Google Scholar] [CrossRef] [PubMed]
- Casarosa, P.; Bakker, R.A.; Verzijl, D.; Navis, M.; Timmerman, H.; Leurs, R.; Smit, M.J. Constitutive signaling of the human cytomegalovirus-encoded chemokine receptor US28. J. Biol. Chem. 2001, 276, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Fraile-Ramos, A.; Kledal, T.N.; Pelchen-Matthews, A.; Bowers, K.; Schwartz, T.W.; Marsh, M. The human cytomegalovirus US28 protein is located in endocytic vesicles and undergoes constitutive endocytosis and recycling. Mol. Biol. Cell 2001, 12, 1737–1749. [Google Scholar] [CrossRef] [PubMed]
- Minisini, R.; Tulone, C.; Lüske, A.; Michel, D.; Mertens, T.; Gierschik, P.; Moepps, B. Constitutive inositol phosphate formation in cytomegalovirus-infected human fibroblasts is due to expression of the chemokine receptor homologue pUS28. J. Virol. 2003, 77, 4489–4501. [Google Scholar] [CrossRef] [PubMed]
- Bakker, R.A.; Casarosa, P.; Timmerman, H.; Smit, M.J.; Leurs, R. Constitutively active Gq/11-coupled receptors enable signaling by co-expressed Gi/o-coupled receptors. J. Biol. Chem. 2004, 279, 5152–5161. [Google Scholar] [CrossRef] [PubMed]
- Neote, K.; DiGregorio, D.; Mak, J.Y.; Horuk, R.; Schall, T.J. Molecular cloning, functional expression, and signaling characteristics of a CC chemokine receptor. Cell 1993, 72, 415–425. [Google Scholar] [CrossRef]
- Maussang, D.; Verzijl, D.; van Walsum, M.; Leurs, R.; Holl, J.; Pleskoff, O.; Michel, D.; van Dongen, G.A.M.S.; Smit, M.J. Human cytomegalovirus-encoded chemokine receptor US28 promotes tumorigenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 13068–13073. [Google Scholar] [CrossRef] [PubMed]
- Moepps, B.; Tulone, C.; Kern, C.; Minisini, R.; Michels, G.; Vatter, P.; Wieland, T.; Gierschik, P. Constitutive serum response factor activation by the viral chemokine receptor homologue pUS28 is differentially regulated by Gαq/11 and Gα16. Cell. Signal. 2008, 20, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Maussang, D.; Langemeijer, E.; Fitzsimons, C.P.; Stigter-van Walsum, M.; Dijkman, R.; Borg, M.K.; Slinger, E.; Schreiber, A.; Michel, D.; Tensen, C.P.; et al. The human cytomegalovirus—Encoded chemokine receptor US28 promotes angiogenesis and tumor formation via cyclooxygenase-2. Cancer Res. 2009, 69, 2861–2869. [Google Scholar] [CrossRef] [PubMed]
- Slinger, E.; Maussang, D.; Schreiber, A.; Siderius, M.; Rahbar, A.; Fraile-Ramos, A.; Lira, S.A.; Soderberg-Naucler, C.; Smit, M.J. HCMV-encoded chemokine receptor US28 mediates proliferative signaling through the IL-6-STAT3 axis. Sci. Signal. 2010, 3, ra58. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Matlaf, L.; Bezrookove, V.; Harkins, L.; Martinez, R.; Greene, M.; Soteropoulos, P.; Cobbs, C.S. Human cytomegalovirus US28 found in glioblastoma promotes an invasive and angiogenic phenotype. Cancer Res. 2011, 71, 6643–6653. [Google Scholar] [CrossRef] [PubMed]
- Langemeijer, E.V.; Slinger, E.; de Munnik, S.; Schreiber, A.; Maussang, D.; Vischer, H.; Verkaar, F.; Leurs, R.; Siderius, M.; Smit, M.J. Constitutive beta-catenin signaling by the viral chemokine receptor US28. PLoS ONE 2012, 7, e48935. [Google Scholar] [CrossRef] [PubMed]
- De Wit, R.H.; Mujić-Delić, A.; van Senten, J.R.; Fraile-Ramos, A.; Siderius, M.; Smit, M.J. Human cytomegalovirus encoded chemokine receptor US28 activates the HIF-1α/PKM2 axis in glioblastoma cells. Oncotarget 2016, 7, 67966–67985. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-E.; Miller, W.E. The HCMV US28 vGPCR induces potent Gαq/PLC-β signaling in monocytes leading to increased adhesion to endothelial cells. Virology 2016, 497, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Vischer, H.F.; Leurs, R.; Smit, M.J. HCMV-encoded G-protein-coupled receptors as constitutively active modulators of cellular signaling networks. Trends. Pharmacol. Sci. 2006, 27, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Waldhoer, M.; Casarosa, P.; Rosenkilde, M.M.; Smit, M.J.; Leurs, R.; Whistler, J.L.; Schwartz, T.W. The carboxyl terminus of human cytomegalovirus-encoded 7 transmembrane receptor US28 camouflages agonism by mediating constitutive endocytosis. J. Biol. Chem. 2003, 278, 19473–19482. [Google Scholar] [CrossRef] [PubMed]
- Fraile-Ramos, A.; Kohout, T.A.; Waldhoer, M.; Marsh, M. Endocytosis of the viral chemokine receptor US28 does not require beta-arrestins but is dependent on the clathrin-mediated pathway. Traffic 2003, 4, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.E.; Houtz, D.A.; Nelson, C.D.; Kolattukudy, P.E.; Lefkowitz, R.J. G-protein-coupled receptor (GPCR) kinase phosphorylation and β-arrestin recruitment regulate the constitutive signaling activity of the human cytomegalovirus US28 GPCR. J. Biol. Chem. 2003, 278, 21663–21671. [Google Scholar] [CrossRef] [PubMed]
- Mokros, T.; Rehm, A.; Droese, J.; Oppermann, M.; Lipp, M.; Höpken, U.E. Surface expression and endocytosis of the human cytomegalovirus-encoded chemokine receptor US28 is regulated by agonist-independent phosphorylation. J. Biol. Chem. 2002, 277, 45122–45128. [Google Scholar] [CrossRef] [PubMed]
- Sherrill, J.D.; Miller, W.E. G protein-coupled receptor (GPCR) kinase 2 regulates agonist-independent Gq/11 signaling from the mouse cytomegalovirus GPCR M33. J. Biol. Chem. 2006, 281, 39796–39805. [Google Scholar] [CrossRef] [PubMed]
- Stropes, M.P.; Schneider, O.D.; Zagorski, W.A.; Miller, J.L.; Miller, W.E. The carboxy terminal tail of HCMV-US28 regulates both chemokine independent and chemokine dependent signaling in HCMV infected cells. J. Virol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Boomker, J.M.; The, T.H.; de Leij, L.F.; Harmsen, M.C. The human cytomegalovirus-encoded receptor US28 increases the activity of the major immediate-early promoter/enhancer. Virus Res. 2006, 118, 196–200. [Google Scholar] [CrossRef] [PubMed]
- DeMeritt, I.B.; Milford, L.E.; Yurochko, A.D. Activation of the NF-κB pathway in human cytomegalovirus-infected cells is necessary for efficient transactivation of the major immediate-early promoter. J. Virol. 2004, 78, 4498–4507. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.-Q.; Zhang, Y.-Y.; Lv, L.-P.; Zhou, X.-P.; Yan, F.; Ma, P.; Xu, J.-B. Human cytomegalovirus-encoded chemokine receptor homolog US28 stimulates the major immediate early gene promoter/enhancer via the induction of CREB. J. Recept. Signal Transduct. Res. 2009, 29, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Frank, T.; Reichel, A.; Larsen, O.; Stilp, A.-C.; Rosenkilde, M.M.; Stamminger, T.; Ozawa, T.; Tschammer, N. Attenuation of chemokine receptor function and surface expression as an immunomodulatory strategy employed by human cytomegalovirus is linked to vGPCR US28. Cell Commun. Signal. 2016, 14, 31. [Google Scholar] [CrossRef] [PubMed]
- Low, H.; Mukhamedova, N.; Cui, H.L.; McSharry, B.P.; Avdic, S.; Hoang, A.; Ditiatkovski, M.; Liu, Y.; Fu, Y.; Meikle, P.J. Cytomegalovirus restructures lipid rafts via a US28/CDC42-mediated pathway, enhancing cholesterol efflux from host cells. Cell Rep. 2016, 16, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Kledal, T.N.; Rosenkilde, M.M.; Schwartz, T.W. Selective recognition of the membrane-bound CX3C chemokine, fractalkine, by the human cytomegalovirus-encoded broad-spectrum receptor US28. FEBS Lett. 1998, 441, 209–214. [Google Scholar] [CrossRef]
- Kuhn, D.E.; Beall, C.J.; Kolattukudy, P.E. The cytomegalovirus US28 protein binds multiple cc chemokines with high affinity. Biochem. Biophys. Res. Commun. 1995, 211, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Miles, T.F.; Spiess, K.; Jude, K.M.; Tsutsumi, N.; Burg, J.S.; Ingram, J.R.; Waghray, D.; Hjorto, G.M.; Larsen, O.; Ploegh, H.L. Viral GPCR US28 can signal in response to chemokine agonists of nearly unlimited structural degeneracy. eLife 2018, 7, e35850. [Google Scholar] [CrossRef] [PubMed]
- Casarosa, P.; Waldhoer, M.; LiWang, P.J.; Vischer, H.F.; Kledal, T.; Timmerman, H.; Schwartz, T.W.; Smit, M.J.; Leurs, R. CC and CX3C chemokines differentially interact with the N terminus of the human cytomegalovirus-encoded US28 receptor. J. Biol. Chem. 2005, 280, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.D.; Clarke, W.P.; von Zastrow, M.; Nichols, D.E.; Kobilka, B.; Weinstein, H.; Javitch, J.A.; Roth, B.L.; Christopoulos, A.; Sexton, P.M.; et al. Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 2007, 320, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zipeto, D.; Bodaghi, B.; Laurent, L.; Virelizier, J.-L.; Michelson, S. Kinetics of transcription of human cytomegalovirus chemokine receptor US28 in different cell types. J. Gen. Virol. 1999, 80, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Paša-Tolić, L.; Wang, D.; Camp, D.G.; Rodland, K.; Wiley, S.; et al. Identification of proteins in human cytomegalovirus (HCMV) particles: The HCMV proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef] [PubMed]
- Bannert, N.; Craig, S.; Farzan, M.; Sogah, D.; Santo, N.V.; Choe, H.; Sodroski, J. Sialylated O-glycans and sulfated tyrosines in the NH2-terminal domain of CC chemokine receptor 5 contribute to high affinity binding of chemokines. J. Exp. Med. 2001, 194, 1661–1674. [Google Scholar] [CrossRef] [PubMed]
- Bodaghi, B.; Jones, T.R.; Zipeto, D.; Vita, C.; Sun, L.; Laurent, L.; Arenzana-Seisdedos, F.; Virelizier, J.L.; Michelson, S. Chemokine sequestration by viral chemoreceptors as a novel viral escape strategy: Withdrawal of chemokines from the environment of cytomegalovirus-infected cells. J. Exp. Med. 1998, 188, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Randolph-Habecker, J.R.; Rahill, B.; Torok-Storb, B.; Vieira, J.; Kolattukudy, P.E.; Rovin, B.H.; Sedmak, D.D. The expression of the cytomegalovirus chemokine receptor homolog US28 sequesters biologically active CC chemokines and alters IL-8 production. Cytokine 2002, 19, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Boomker, J.M.; de Jong, E.K.; de Leij, L.F.; Harmsen, M.C. Chemokine scavenging by the human cytomegalovirus chemokine decoy receptor US28 does not inhibit monocyte adherence to activated endothelium. Antiviral Res. 2006, 69, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Dupont, L.; Reeves, M.B. Cytomegalovirus latency and reactivation: Recent insights into an age old problem. Rev. Med. Virol. 2015, 26, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E.; Sedikides, G.X.; Mason, G.M.; Okecha, G.; Wills, M.R. Human cytomegalovirus (HCMV)-specific CD4+ T cells are polyfunctional and can respond to HCMV-infected dendritic cells in vitro. J. Virol. 2017, 91, e02128-16. [Google Scholar] [CrossRef] [PubMed]
- Beisser, P.S.; Laurent, L.; Virelizier, J.L.; Michelson, S. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J. Virol. 2001, 75, 5949–5957. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K.; Abendroth, A.; Cunningham, A.L.; Slobedman, B. Viral gene expression during the establishment of human cytomegalovirus latent infection in myeloid progenitor cells. Blood 2006, 108, 3691–3699. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.; Jordan, C.T.; Terhune, S.S.; High, K.; Shenk, T. Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood 2004, 104, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Walther, A.; Raven, K.; Benedict, C.A.; Mason, G.M.; Sinclair, J. The myeloid transcription factor GATA-2 regulates the viral UL144 gene during human cytomegalovirus latency in an isolate-specific manner. J. Virol. 2013, 87, 4261–4271. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Tarrant-Elorza, M.; Pari, G.S. Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14 (+) monocytes and CD34 (+) cells. PLoS Pathog. 2013, 9, e1003366. [Google Scholar] [CrossRef] [PubMed]
- Cardin, R.D.; Schaefer, G.C.; Allen, J.R.; Davis-Poynter, N.J.; Farrell, H.E. The M33 chemokine receptor homolog of murine cytomegalovirus exhibits a differential tissue-specific role during in vivo replication and latency. J. Virol. 2009, 83, 7590–7601. [Google Scholar] [CrossRef] [PubMed]
- Bittencourt, F.M.; Wu, S.E.; Bridges, J.P.; Miller, W.E. The M33 G protein-coupled receptor encoded by murine cytomegalovirus is dispensable for hematogenous dissemination but is required for growth within the salivary gland. J. Virol. 2014, 88, 11811–11824. [Google Scholar] [CrossRef] [PubMed]
- Beisser, P.S.; Grauls, G.; Bruggeman, C.A.; Vink, C. Deletion of the R78 G protein-coupled receptor gene from rat cytomegalovirus results in an attenuated, syncytium-inducing mutant strain. J. Virol. 1999, 73, 7218–7230. [Google Scholar] [PubMed]
- Krishna, B.A.; Humby, M.S.; O’Connor, C.M. US28 mediated attenuation of FOS signaling to the MIEP is important for the establishment of HCMV latency. In 43rd Annual International Herpesvirus Workshop Abstract Book, Proceedings of the 43rd Annual International Herpesvirus Workshop, Vancouver, BC, Canada, 2018; Abstract #4.03; IHW: Vancouver, BC, Canada, 2018. [Google Scholar]
- Zhu, D.; Pan, C.; Sheng, J.; Liang, H.; Bian, Z.; Liu, Y.; Trang, P.; Wu, J.; Liu, F.; Zhang, C.-Y. Human cytomegalovirus reprogrammes haematopoietic progenitor cells into immunosuppressive monocytes to achieve latency. Nat. Microbiol. 2018, 3, 503. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Humby, M.S.; Wu, S.-E.; Miller, W.E.; O’Connor, C.M. US28 mediated attenuation of FOS signaling to the MIEP is important for the establishment and maintenance of HCMV latency. p status. (unpublished; manuscript in preparation).
- Samanta, M.; Harkins, L.; Klemm, K.; Britt, W.J.; Cobbs, C.S. High prevalence of human cytomegalovirus in prostatic intraepithelial neoplasia and prostatic carcinoma. J. Urol. 2003, 170, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Yan, Q.; Wang, Z.; Chen, X.; Zhang, X.; Guo, Y.; Li, J.J. Human cytomegalovirus in neoplastic cells of epstein-barr virus negative hodgkin’s disease. Int. J. Oncol. 2002, 21, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Harkins, L.; Volk, A.L.; Samanta, M.; Mikolaenko, I.; Britt, W.J.; Bland, K.I.; Cobbs, C.S. Specific localisation of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet 2002, 360, 1557–1563. [Google Scholar] [CrossRef]
- Cobbs, C.S.; Harkins, L.; Samanta, M.; Gillespie, G.Y.; Bharara, S.; King, P.H.; Nabors, L.B.; Cobbs, C.G.; Britt, W.J. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002, 62, 3347–3350. [Google Scholar] [PubMed]
- Mitchell, D.A.; Xie, W.; Schmittling, R.; Learn, C.; Friedman, A.; McLendon, R.E.; Sampson, J.H. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol. 2008, 10, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Scheurer, M.E.; Bondy, M.L.; Aldape, K.D.; Albrecht, T.; El-Zein, R. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathol. 2008, 116, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Baryawno, N.; Rahbar, A.; Wolmer-Solberg, N.; Taher, C.; Odeberg, J.; Darabi, A.; Khan, Z.; Sveinbjörnsson, B.; FuskevÅg, O.-M.; Segerström, L. Detection of human cytomegalovirus in medulloblastomas reveals a potential therapeutic target. J. Clin. Investig. 2011, 121, 4043–4055. [Google Scholar] [CrossRef] [PubMed]
- Price, R.L.; Harkins, L.; Chiocca, E.A.; Zhang, P.J.; Kurt, H.; Iwenofu, O.H. Human cytomegalovirus is present in alveolar soft part sarcoma. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Dziurzynski, K.; Chang, S.M.; Heimberger, A.B.; Kalejta, R.F.; McGregor Dallas, S.R.; Smit, M.; Soroceanu, L.; Cobbs, C.S. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol. 2012, 14, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E. Gammaherpesviruses and lymphoproliferative disorders. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 who classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Baumgarten, P.; Mittelbronn, M.; Driever, P.H.; Doerr, H.W.; Cinatl, J. Oncomodulation by human cytomegalovirus: Novel clinical findings open new roads. Med. Microbiol. Immunol. 2011, 200, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, A.; Stragliotto, G.; Orrego, A.; Peredo, I.; Taher, C.; Willems, J.; Söderberg-Naucler, C. Low levels of human cytomegalovirus infection in glioblastoma multiforme associates with patient survival; -a case-control study. Herpesviridae 2012, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Söderberg-Nauclér, C.; Rahbar, A.; Stragliotto, G. Survival in patients with glioblastoma receiving valganciclovir. N. Engl. J. Med. 2013, 369, 985–986. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Wang, J.; Tanksley, J.P.; Mobley, B.C.; Ayers, G.D.; Moots, P.L.; Clark, S.W. Valganciclovir and bevacizumab for recurrent glioblastoma: A single-institution experience. Mol. Clin. Oncol. 2016, 4, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.-W.; Hellstrand, K.; Larsson, E. Absence of cytomegalovirus in high-coverage DNA sequencing of human glioblastoma multiforme. Int. J. Cancer. 2015, 136, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.S.; Abdalhamid, B.A.; El-Badawy, S.A.; Sorour, Y.M.; Almsned, F.M.; Al-Abbadi, M.A. Expression of cytomegalovirus in glioblastoma multiforme: Myth or reality? Br. J. Neurosurg. 2016, 30, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Baumgarten, P.; Michaelis, M.; Rothweiler, F.; Starzetz, T.; Rabenau, H.F.; Berger, A.; Jennewein, L.; Braczynski, A.K.; Franz, K.; Seifert, V. Human cytomegalovirus infection in tumor cells of the nervous system is not detectable with standardized pathologico-virological diagnostics. Neuro. Oncol. 2014, 16, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.-W.; Alaei-Mahabadi, B.; Samuelsson, T.; Lindh, M.; Larsson, E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat. Commun. 2013, 4, 2513. [Google Scholar] [CrossRef] [PubMed]
- Hellstrand, K.; Martner, A.; Bergstrom, T. Valganciclovir in patients with glioblastoma. N. Engl. J. Med. 2013, 369, 2066–2067. [Google Scholar] [PubMed]
- dos Santos, C.J.; Stangherlin, L.M.; Figueiredo, E.G.; Corrêa, C.; Teixeira, M.J.; da Silva, M.C.C. High prevalence of HCMV and viral load in tumor tissues and peripheral blood of glioblastoma multiforme patients. J. Med. Virol. 2014, 86, 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Akhavan, A.; Cobbs, C.S. Platelet-derived growth factor-α receptor activation is required for human cytomegalovirus infection. Nature 2008, 455, 391. [Google Scholar] [CrossRef] [PubMed]
- Toepoel, M.; Joosten, P.H.; Knobbe, C.B.; Afink, G.B.; Zotz, R.B.; Steegers-Theunissen, R.P.; Reifenberger, G.; van Zoelen, E.J. Haplotype-specific expression of the human PDGFRA gene correlates with the risk of glioblastomas. Int. J. Cancer 2008, 123, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Dey, M.; Ahmed, A.U.; Lesniak, M.S. Cytomegalovirus and glioma: Putting the cart before the horse. J. Neurol. Neurosurg. Psychiatry 2015, 86, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Stragliotto, G.; Rahbar, A.; Solberg, N.W.; Lilja, A.; Taher, C.; Orrego, A.; Bjurman, B.; Tammik, C.; Skarman, P.; Peredo, I. Effects of valganciclovir as an add-on therapy in patients with cytomegalovirus-positive glioblastoma: A randomized, double-blind, hypothesis-generating study. Int. J. Cancer 2013, 133, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Hortal, A.M.; Vermeulen, J.F.; Van Hecke, W.; Bovenschen, N. Oncogenic role of cytomegalovirus in medulloblastoma? Cancer Lett. 2017, 408, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Kumar, A. The oncogenic potential of human cytomegalovirus and breast cancer. Front. Oncol. 2014, 4, 230. [Google Scholar] [CrossRef] [PubMed]
- Heukers, R.; Fan, T.; De, T.G.; Bebelman, M.; Lagerweij, T.; Vieira, J.; Smits-de, L.V.; Rahbar, A.; Söderberg-Naucler, C.; Würdinger, T. The constitutive activity of the virally encoded chemokine receptor US28 accelerates glioblastoma growth. Oncogene 2018, 37, 4110–4121. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, B.; Renzette, N.; Kowalik, T.F. Genetic analysis of cytomegalovirus in malignant gliomas. J. Virol. 2012, 86, 6815–6824. [Google Scholar] [CrossRef] [PubMed]
- Dziurzynski, K.; Wei, J.; Qiao, W.; Hatiboglu, M.A.; Kong, L.-Y.; Wu, A.; Wang, Y.; Cahill, D.; Levine, N.; Prabhu, S. Glioma-associated cytomegalovirus mediates subversion of the monocyte lineage to a tumor propagating phenotype. Clin. Cancer Res. 2011, 17, 4642–4649. [Google Scholar] [CrossRef] [PubMed]
- de la Iglesia, N.; Konopka, G.; Lim, K.-L.; Nutt, C.L.; Bromberg, J.F.; Frank, D.A.; Mischel, P.S.; Louis, D.N.; Bonni, A. Deregulation of a STAT3–interleukin 8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J. Neurosci. 2008, 28, 5870–5878. [Google Scholar] [CrossRef] [PubMed]
- de la Iglesia, N.; Konopka, G.; Puram, S.V.; Chan, J.A.; Bachoo, R.M.; You, M.J.; Levy, D.E.; DePinho, R.A.; Bonni, A. Identification of a pten-regulated stat3 brain tumor suppressor pathway. Genes Dev. 2008, 22, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lathia, J.D.; Wu, Q.; Wang, J.; Li, Z.; Heddleston, J.M.; Eyler, C.E.; Elderbroom, J.; Gallagher, J.; Schuschu, J. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells 2009, 27, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Vischer, H.F.; Siderius, M.; Leurs, R.; Smit, M.J. Herpesvirus-encoded GPCRs: Neglected players in inflammatory and proliferative diseases? Nat. Rev. Drug Discov. 2014, 13, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Price, R.L.; Song, J.; Bingmer, K.; Kim, T.H.; Yi, J.-Y.; Nowicki, M.O.; Mo, X.; Hollon, T.; Murnan, E.; Alvarez-Breckenridge, C.; et al. Cytomegalovirus contributes to glioblastoma in the context of tumor suppressor mutations. Cancer Res. 2013, 73, 3441–3450. [Google Scholar] [CrossRef] [PubMed]
- Bongers, G.; Maussang, D.; Muniz, L.R.; Noriega, V.M.; Fraile-Ramos, A.; Barker, N.; Marchesi, F.; Thirunarayanan, N.; Vischer, H.F.; Qin, L.; et al. The cytomegalovirus-encoded chemokine receptor US28 promotes intestinal neoplasia in transgenic mice. J. Clin. Investig. 2010, 120, 3969–3978. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.-Z.; Xu, J.-G.; Zhou, Y.-H.; Zheng, J.-H.; Lin, K.-Z.; Zheng, S.-Z.; Ye, M.-S.; He, Y.; Liu, C.-B.; Xue, Z.-X. Human cytomegalovirus-encoded US28 may act as a tumor promoter in colorectal cancer. World J. Gastroenterol. 2016, 22, 2789. [Google Scholar] [CrossRef] [PubMed]
- Streblow, D.N.; Dumortier, J.; Moses, A.V.; Orloff, S.L.; Nelson, J.A. Mechanisms of cytomegalovirus-accelerated vascular disease: Induction of paracrine factors that promote angiogenesis and wound healing. Curr. Top. Microbiol. Immunol. 2008, 325, 397–415. [Google Scholar] [PubMed]
- Wang, H.; Peng, G.; Bai, J.; He, B.; Huang, K.; Hu, X.; Liu, D. Cytomegalovirus infection and relative risk of cardiovascular disease (ischemic heart disease, stroke, and cardiovascular death): A meta-analysis of prospective studies up to 2016. J. Am. Heart Assoc. 2017, 6, e005025. [Google Scholar] [CrossRef] [PubMed]
- Lamon, B.D.; Hajjar, D.P. Inflammation at the molecular interface of atherogenesis: An anthropological journey. Am. J. Pathol. 2008, 173, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atheroschlerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Muhlestein, J.B.; Horne, B.D.; Carlquist, J.F.; Madsen, T.E.; Bair, T.L.; Pearson, R.R.; Anderson, J.L. Cytomegalovirus seropositivity and c-reactive protein have independent and combined predictive value for mortality in patients with angiographically demonstrated coronary artery disease. Circulation 2000, 102, 1917–1923. [Google Scholar] [CrossRef] [PubMed]
- Söderberg-Nauclér, C. Does cytomegalovirus play a causative role in the development of various inflammatory diseases and cancer? J. Intern. Med. 2006, 259, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Melnick, J.L.; Dreesman, G.R.; McCollum, C.H.; Petrie, B.L.; Burek, J.; DeBakey, M.E. Cytomegalovirus antigen within human arterial smooth muscle cells. Lancet 1983, 322, 644–647. [Google Scholar] [CrossRef]
- Pampou, S.Y.; Gnedoy, S.N.; Bystrevskaya, V.B.; Smirnov, V.N.; Chazov, E.I.; Melnick, J.L.; DeBakey, M.E. Cytomegalovirus genome and the immediate-early antigen in cells of different layers of human aorta. Virchows Arch. 2000, 436, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Holm, P.W.; Slart, R.H.; Zeebregts, C.J.; Hillebrands, J.L.; Tio, R.A. Atherosclerotic plaque development and instability: A dual role for VEGF. Ann. Med. 2009, 41, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, J.; Streblow, D.N.; Moses, A.V.; Jacobs, J.M.; Kreklywich, C.N.; Camp, D.; Smith, R.D.; Orloff, S.L.; Nelson, J.A. Human cytomegalovirus secretome contains factors that induce angiogenesis and wound healing. J. Virol. 2008, 82, 6524–6535. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Juss, J.K.; Krishna, B.; Herre, J.; Chilvers, E.R.; Sinclair, J. Alveolar macrophages isolated directly from human cytomegalovirus (HCMV)—Seropositive individuals are sites of HCMV reactivation in vivo. J. Infect. Dis. 2015, 211, 1936–1942. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.; Sissons, P.; Sinclair, J. Reactivation of human cytomegalovirus in dendritic cells. Discov. Med. 2005, 5, 170–174. [Google Scholar] [PubMed]
- Taylor-Wiedeman, J.; Sissons, P.; Sinclair, J. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J. Virol. 1994, 68, 1597–1604. [Google Scholar] [PubMed]
- Lee, S.; Chung, Y.H.; Lee, C. Us28, a virally-encoded GPCR as an antiviral target for human cytomegalovirus infection. Biomol. Ther. 2017, 25, 69. [Google Scholar] [CrossRef] [PubMed]
- Vischer, H.F.; Hulshof, J.W.; Hulscher, S.; Fratantoni, S.A.; Verheij, M.H.P.; Victorina, J.; Smit, M.J.; de Esch, I.J.P.; Leurs, R. Identification of novel allosteric nonpeptidergic inhibitors of the human cytomegalovirus-encoded chemokine receptor US28. Bioorg. Med. Chem. 2010, 18, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Hesselgesser, J.; Ng, H.P.; Liang, M.; Zheng, W.; May, K.; Bauman, J.G.; Monahan, S.; Islam, I.; Wei, G.P.; Ghannam, A. Identification and characterization of small molecule functional antagonists of the CCR1 chemokine receptor. J. Biol. Chem. 1998, 273, 15687–15692. [Google Scholar] [CrossRef] [PubMed]
- Hulshof, J.W.; Vischer, H.F.; Verheij, M.H.P.; Fratantoni, S.A.; Smit, M.J.; de Esch, I.J.P.; Leurs, R. Synthesis and pharmacological characterization of novel inverse agonists acting on the viral-encoded chemokine receptor US28. Bioorg. Med. Chem. 2006, 14, 7213–7230. [Google Scholar] [CrossRef] [PubMed]
- Hulshof, J.W.; Casarosa, P.; Menge, W.M.P.B.; Kuusisto, L.M.S.; van der Goot, H.; Smit, M.J.; de Esch, I.J.P.; Leurs, R. Synthesis and structure-activity relationship of the first nonpeptidergic inverse agonists for the human cytomegalovirus encoded chemokine receptor US28. J. Med. Chem. 2005, 48, 6461–6471. [Google Scholar] [CrossRef] [PubMed]
- Kralj, A.; Wetzel, A.; Mahmoudian, S.; Stamminger, T.; Tschammer, N.; Heinrich, M.R. Identification of novel allosteric modulators for the G-protein coupled US28 receptor of human cytomegalovirus. Bioorg. Med. Chem. Lett. 2011, 21, 5446–5450. [Google Scholar] [CrossRef] [PubMed]
- Kralj, A.; Nguyen, M.-T.; Tschammer, N.; Ocampo, N.; Gesiotto, Q.; Heinrich, M.R.; Phanstiel Iv, O. Development of flavonoid-based inverse agonists of the key signaling receptor US28 of human cytomegalovirus. J. Med. Chem. 2013, 56, 5019–5032. [Google Scholar] [CrossRef] [PubMed]
- Kralj, A.; Kurt, E.; Tschammer, N.; Heinrich, M.R. Synthesis and biological evaluation of biphenyl amides that modulate the US28 receptor. Chem. Med. Chem. 2014, 9, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Amărandi, R.-M.; Lückmann, M.; Melynis, M.; Jakobsen, M.H.; Fallah, Z.; Spiess, K.; Hjortø, G.M.; Pui, A.; Frimurer, T.M.; Rosenkilde, M.M. Ligand-selective small molecule modulators of the constitutively active vGPCR US28. Eur. J. Med. Chem. 2018, 15, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Tschammer, N. Allosteric modulation of the G protein-coupled US28 receptor of human cytomegalovirus: Are the small-weight inverse agonist of US28 ‘camouflaged’agonists? Bioorg. Med. Chem. Lett. 2014, 24, 3744–3747. [Google Scholar] [CrossRef] [PubMed]
- Spiess, K.; Jeppesen, M.G.; Malmgaard-Clausen, M.; Krzywkowski, K.; Dulal, K.; Cheng, T.; Hjortø, G.M.; Larsen, O.; Burg, J.S.; Jarvis, M.A.; et al. Rationally designed chemokine-based toxin targeting the viral G protein-coupled receptor US28 potently inhibits cytomegalovirus infection in vivo. Proc. Natl. Acad. Sci. USA 2015, 112, 8427–8432. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Spiess, K.; Poole, E.L.; Lau, B.; Voigt, S.; Kledal, T.N.; Rosenkilde, M.M.; Sinclair, J.H. Targeting the latent cytomegalovirus reservoir with an antiviral fusion toxin protein. Nat. Commun. 2017, 8, 14321. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Hieshima, K.; Haskell, C.; Baba, M.; Nagira, M.; Nishimura, M.; Kakizaki, M.; Takagi, S.; Nomiyama, H.; Schall, T.J.; et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 1997, 91, 521–530. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HCMV-Encoded GPCR | Function in Lytic Infection of Fibroblasts | Function in Lytic Infection of Other Cells | Function in Latency | Interactions with Other GPCRS |
|---|---|---|---|---|
| US27 | Extracellular virus spread [47] | Dispensable for viral growth in epithelial cells; required for extracellular spread in endothelial cells [47] | Not expressed [52,53] | Enhances CXCR4 activation [42,50]. Heteromerizes with pUS28 [54] |
| US28 | Dispensable [46,48,55] | Augments infection of epithelial cells [48]; dispensable for growth in endothelial cells [55] | Required for latency [52,56] | Heteromerizes with pUS27, pUL33, and pUL78. Interaction with either pUL33 or pUL78 abrogates pUS28-mediated NFκB activation [54] |
| UL33 | Dispensable [40] | Unknown | Detected [53]; unknown function | Heteromerizes with pUS28 and abrogates pUS28-mediated NFκB activation [54]. Binds CCR5 or CXCR4 to reduce signaling activity in both [41]. |
| UL78 | Dispensable [44,45] | Efficient viral replication in endothelial & epithelial cells. Efficient entry & virion delivery into epithelial cells [45]. | Detected [53]; unknown function | Heteromerizes with pUS28 and abrogates pUS28-mediated NFκB activation [54]. Binds CCR5 to increase signaling activity [41]. Binds CXCR4 to reduce signaling activity [41]. |
| Cell System | Ligands | Phenotypic Change |
|---|---|---|
| K562 ⌘f | CCL2 and CCL5 | Calcium release [59] |
| HEK293T § & infected HUVECs g | CCL7 and CCL5 | Calcium release and MAP kinase via Gα16 [60] |
| Infected fibroblast | CCL2 and CCL5 | Calcium release [46] |
| Infected arterial SMCs | CCL5 (inhibited by CX3CL1) | Chemotaxis via Gα12/13 [61,62,63,64] |
| Cos-7 cells § | Constitutive, CX3CL1 inhibits | PLC, NFκB via Gαq [65,66] |
| Infected fibroblasts | Constitutive | PLC via Gαq [67] |
| Cos-7 cells § | Constitutive, antagonized by CCL5 | PLC and NFκB via Gαo and Gαq11 [68] |
| HEK293T § | Constitutive | CREB/NFAT a via Gαq [69] |
| NIH-3T3 ⌘h & infected U373 | Constitutive | VEGF b secretion via Gαq and MAPK [70] |
| Cos-7 cells § | Constitutive | Serum response factor via Gαo and Gαq11 (inhibited by Gα16) [71] |
| Mouse macrophages ⌘ | CX3CL1 (inhibited by CCL5) | Chemotaxis via Gαq [64] |
| NIH-3T3 § & infected fibroblasts | Constitutive | COX2 and VEGF via Gαq; NFκB [72] |
| NIH-3T3 § & HEK293T §; infected U373MG | Constitutive | NFκB induction of IL6, VEGF secretion inducing JAK c/STAT3 [73] |
| Non-proliferating hippocampal cells ¶ & HUVECs ¶ | CCL5 | “Invasive phenotypes” via STAT3, AKT d, ERK1/2, FAK, Src, and eNOS [74] |
| NIH-3T3 § & HEK293T §; infected fibroblasts & U373MG | Constitutive | β-catenin via both Gα12 and Gαq together [75] |
| Infected HASMC, U373MG, HFFs, & HUVECs | Constitutive | PLC-β via Gαq and Gα11 in all cell types tested [55] |
| Infected HASMC & HFFs | CCL5 | Calcium release via Gα12/13 [55] |
| U251 ¶ & NIH-3T3 ⌘ | Constitutive | VEGF secretion and HIF1-α activation with Akt and PKM2 e [76] |
| THP-1 cells ¶ | Constitutive | PLC-β via Gαq [77] |
| THP-1 cells ⌘ | Constitutive | Attenuation of MAPK and NFκB [56] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishna, B.A.; Miller, W.E.; O’Connor, C.M. US28: HCMV’s Swiss Army Knife. Viruses 2018, 10, 445. https://doi.org/10.3390/v10080445
Krishna BA, Miller WE, O’Connor CM. US28: HCMV’s Swiss Army Knife. Viruses. 2018; 10(8):445. https://doi.org/10.3390/v10080445
Chicago/Turabian StyleKrishna, Benjamin A., William E. Miller, and Christine M. O’Connor. 2018. "US28: HCMV’s Swiss Army Knife" Viruses 10, no. 8: 445. https://doi.org/10.3390/v10080445
APA StyleKrishna, B. A., Miller, W. E., & O’Connor, C. M. (2018). US28: HCMV’s Swiss Army Knife. Viruses, 10(8), 445. https://doi.org/10.3390/v10080445