Unprecedented Diversity of ssDNA Phages from the Family Microviridae Detected within the Gut of a Protochordate Model Organism (Ciona robusta)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collections and Library Preparations
2.2. Identification and Annotation of Microviridae Genomes
2.3. Genome Comparisons and Phylogenetic Analysis
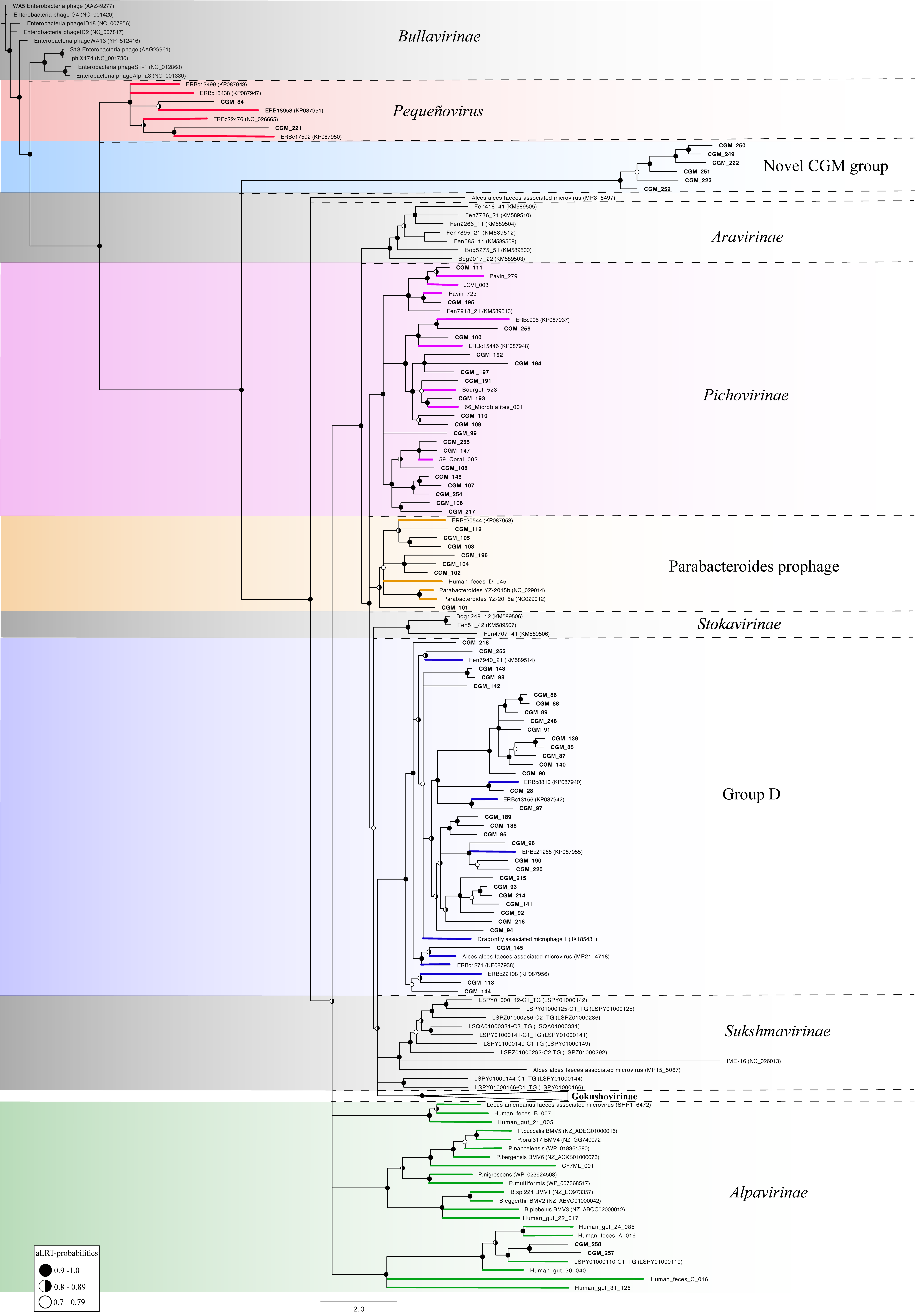
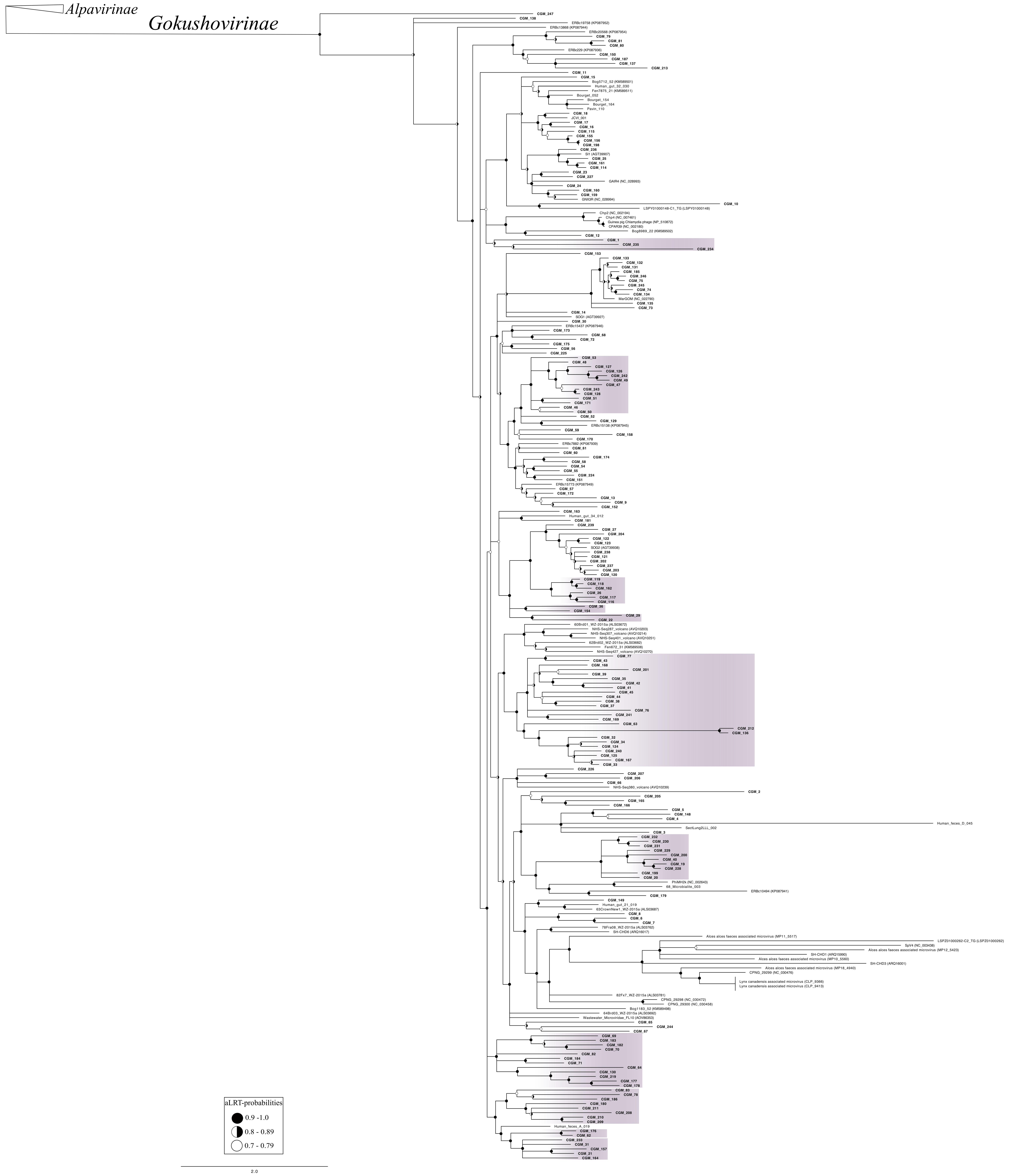
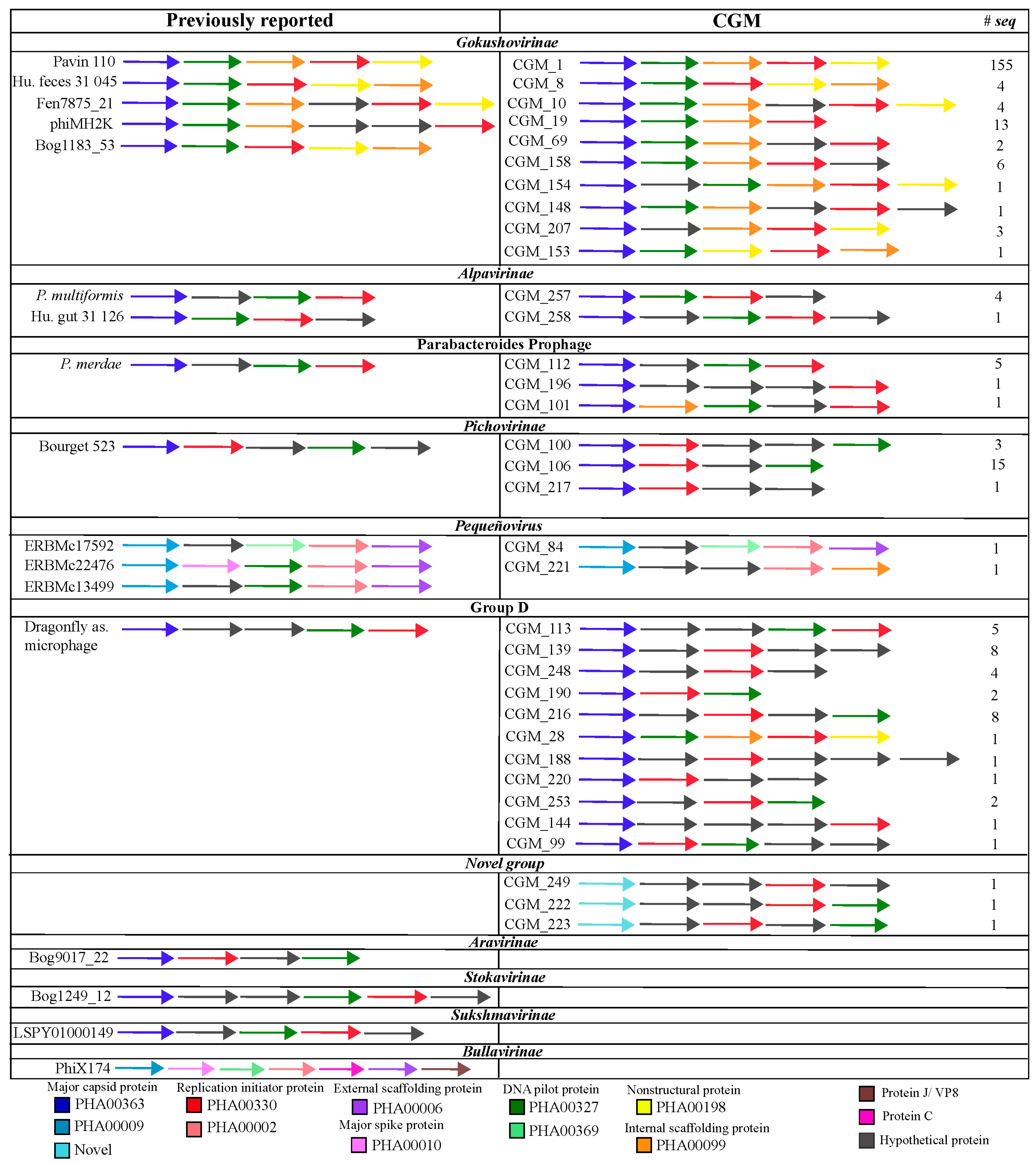
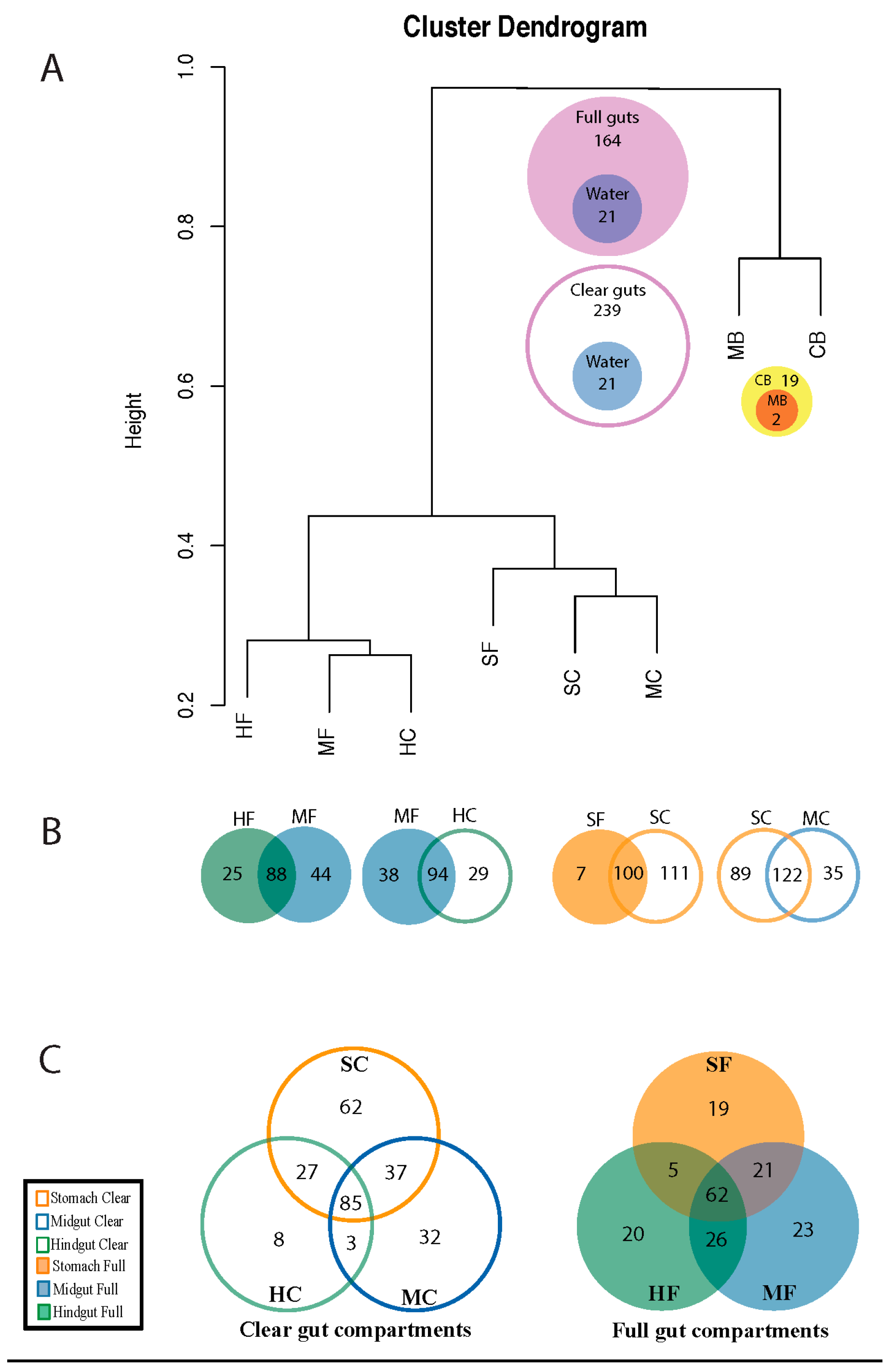
3. Results
3.1. Diversity of Microviridae in Ciona Gut Compartments
3.2. Structure of Microviridae Communities
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Theis, K.R.; Dheilly, N.M.; Klassen, J.L.; Brucker, R.M.; Baines, J.F.; Bosch, T.C.; Cryan, J.F.; Gilbert, S.F.; Goodnight, C.J.; Lloyd, E.A. Getting the hologenome concept right: An eco-evolutionary framework for hosts and their microbiomes. mSystems 2016, 1, e00028-16. [Google Scholar] [CrossRef] [PubMed]
- Bordenstein, S.R.; Theis, K.R. Host biology in light of the microbiome: Ten principles of holobionts and hologenomes. PLoS Biol. 2015, 13, e1002226. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Hewson, I.; Felts, B.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol. 2003, 185, 6220–6223. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.K.; Maurice, C.F. Ménage à trois in the human gut: Interactions between host, bacteria and phages. Nat. Rev. Microbiol. 2017, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Manrique, P.; Dills, M.; Young, M.J. The human gut phage community and its implications for health and disease. Viruses 2017, 9, 141. [Google Scholar] [CrossRef] [PubMed]
- Manrique, P.; Bolduc, B.; Walk, S.T.; van der Oost, J.; de Vos, W.M.; Young, M.J. Healthy human gut phageome. Proc. Natl. Acad. Sci. USA 2016, 113, 10400–10405. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Makarova, K.S.; Gussow, A.B.; Krupovic, M.; Segall, A.; Edwards, R.A.; Koonin, E.V. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol. 2018, 3, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Park, E.-J.; Roh, S.W.; Bae, J.-W. Diversity and abundance of single-stranded DNA viruses in human feces. Appl. Environ. Microbiol. 2011, 77, 8062–8070. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Koskella, B.; Brockhurst, M.A. Bacteria–phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 2014, 38, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, O.H.; Kushmaro, A.; Brenner, A. Bacteriophage predation regulates microbial abundance and diversity in a full-scale bioreactor treating industrial wastewater. ISME J. 2010, 4, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Thurber, R.V. Viruses manipulate the marine environment. Nature 2009, 459, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M. Marine viruses: Truth or dare. Annu. Rev. Mar. Sci. 2012, 4, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, E.; Ianiro, G.; Attili, F.; Bassanelli, C.; De Santis, A.; Gasbarrini, A. The human gut microbiota and virome: Potential therapeutic implications. Dig. Liver Dis. 2015, 47, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Thurber, R.V.; Payet, J.P.; Thurber, A.R.; Correa, A.M. Virus–host interactions and their roles in coral reef health and disease. Nat. Rev. Microbiol. 2017, 15, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Dishaw, L.J.; Flores-Torres, J.; Lax, S.; Gemayel, K.; Leigh, B.; Melillo, D.; Mueller, M.G.; Natale, L.; Zucchetti, I.; De Santis, R.; et al. The gut of geographically disparate Ciona intestinalis harbors a core microbiota. PLoS ONE 2014, 9, e93386. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.G.; Beichman, A.C.; Roman, J.; Scott, J.J.; Emerson, D.; McCarthy, J.J.; Girguis, P.R. Baleen whales host a unique gut microbiome with similarities to both carnivores and herbivores. Nat. Commun. 2015, 6, 8285. [Google Scholar] [CrossRef] [PubMed]
- Soverini, M.; Quercia, S.; Biancani, B.; Furlati, S.; Turroni, S.; Biagi, E.; Consolandi, C.; Peano, C.; Severgnini, M.; Rampelli, S. The bottlenose dolphin (Tursiops truncatus) faecal microbiota. FEMS Microbiol. Ecol. 2016, 92, fiw055. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, R.; Kim, H.B. The intestinal microbiome of the pig. Anim. Health Res. Rev. 2012, 13, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Laffy, P.W.; Wood-Charlson, E.M.; Turaev, D.; Jutz, S.; Pascelli, C.; Botté, E.S.; Bell, S.C.; Peirce, T.E.; Weynberg, K.D.; van Oppen, M.J. Reef invertebrate viromics: Diversity, host specificity and functional capacity. Environ. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, L.A.; Jones, B.V. The human gut virome: A multifaceted majority. Front. Microbiol. 2015, 6, 918. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.R.; Davis, N.; Hoyles, L. The human intestinal virome in health and disease. Aliment. Pharmacol. Ther. 2017, 46, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.; Shanahan, F.; Stanton, C.; Hill, C.; Coffey, A.; Ross, R.P. Movers and shakers: Influence of bacteriophages in shaping the mammalian gut microbiota. Gut Microbes 2013, 4, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Metzger, R.N.; Krug, A.B.; Eisenächer, K. Enteric virome sensing—Its role in intestinal homeostasis and immunity. Viruses 2018, 10, 146. [Google Scholar] [CrossRef] [PubMed]
- Keen, E.C.; Dantas, G. Close encounters of three kinds: Bacteriophages, commensal bacteria, and host immunity. Trends Microbiol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, P.W.; Jeffery, I.B. Gut microbiota and aging. Science 2015, 350, 1214–1215. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Blanton, L.V.; Cao, S.; Zhao, G.; Manary, M.; Trehan, I.; Smith, M.I.; Wang, D.; Virgin, H.W.; Rohwer, F.; et al. Gut DNA viromes of Malawian twins discordant for severe acute malnutrition. Proc. Natl. Acad. Sci. USA 2015, 112, 11941–11946. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Solonenko, N.E.; Dang, V.T.; Poulos, B.T.; Schwenck, S.M.; Goldsmith, D.B.; Coleman, M.L.; Breitbart, M.; Sullivan, M.B. Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 2016, 4, e2777. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Hua, X.; Zhang, W.; Yang, S.; Shen, Q.; Hu, H.; Li, J.; Liu, Z.; Wang, X.; Wang, H.; et al. Viral metagenomics analysis of feces from coronary heart disease patients reveals the genetic diversity of the Microviridae. Virol. Sin. 2017, 2, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Forterre, P. Microviridae goes temperate: Microvirus-related proviruses reside in the genomes of Bacteroidetes. PLoS ONE 2011, 6, e19893. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Krupovic, M.; Poulet, A.; Debroas, D.; Enault, F. Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS ONE 2012, 7, e40418. [Google Scholar] [CrossRef] [PubMed]
- Waller, A.S.; Yamada, T.; Kristensen, D.M.; Kultima, J.R.; Sunagawa, S.; Koonin, E.V.; Bork, P. Classification and quantification of bacteriophage taxa in human gut metagenomes. ISME J. 2014, 8, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Wu, M.; McNulty, N.P.; Rohwer, F.L.; Gordon, J.I. Gnotobiotic mouse model of phage–bacterial host dynamics in the human gut. Proc. Natl. Acad. Sci. USA 2013, 110, 20236–20241. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.S.; Wagner, J.; Mansfield, C.S.; Stevens, M.; Gilkerson, J.R.; Kirkwood, C.D. Characterisation of the canine faecal virome in healthy dogs and dogs with acute diarrhoea using shotgun metagenomics. PLoS ONE 2017, 12, e0178433. [Google Scholar] [CrossRef] [PubMed]
- Tikhe, C.V.; Husseneder, C. Metavirome sequencing of the termite gut reveals the presence of an unexplored bacteriophage community. Front. Microbiol. 2018, 8, 2548. [Google Scholar] [CrossRef] [PubMed]
- D’arc, M.; Furtado, C.; Siqueira, J.D.; Seuánez, H.N.; Ayouba, A.; Peeters, M.; Soares, M.A. Assessment of the gorilla gut virome in association with natural simian immunodeficiency virus infection. Retrovirology 2018, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Székely, A.J.; Breitbart, M. Single-stranded DNA phages: From early molecular biology tools to recent revolutions in environmental microbiology. FEMS Microbiol. Lett. 2016, 363, fnw027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, L.; Deng, X.; Kapusinszky, B.; Pesavento, P.A.; Delwart, E. Faecal virome of cats in an animal shelter. J. Gen. Virol. 2014, 95, 2553–2564. [Google Scholar] [CrossRef] [PubMed]
- Smits, S.L.; Raj, V.S.; Oduber, M.D.; Schapendonk, C.M.; Bodewes, R.; Provacia, L.; Stittelaar, K.J.; Osterhaus, A.D.; Haagmans, B.L. Metagenomic analysis of the ferret fecal viral flora. PLoS ONE 2013, 8, e71595. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shan, T.; Wang, C.; Côté, C.; Kolman, J.; Onions, D.; Gulland, F.M.; Delwart, E. The fecal viral flora of California sea lions. J. Virol. 2011, 85, 9909–9917. [Google Scholar] [CrossRef] [PubMed]
- Van den Brand, J.M.; van Leeuwen, M.; Schapendonk, C.M.; Simon, J.H.; Haagmans, B.L.; Osterhaus, A.D.; Smits, S.L. Metagenomic analysis of the viral flora of pine marten and European badger feces. J. Virol. 2011, 86, 2360–2365. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Haynes, M.; Kelley, S.; Angly, F.; Edwards, R.A.; Felts, B.; Mahaffy, J.M.; Mueller, J.; Nulton, J.; Rayhawk, S.; et al. Viral diversity and dynamics in an infant gut. Res. Microbiol. 2008, 159, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.S.; Wang, D.; Holtz, L.R. The bacterial microbiome and virome milestones of infant development. Trends Microbiol. 2016, 24, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.; Handley, S.; Baldridge, M.; Droit, L.; Liu, C.; Keller, B.; Kambal, A.; Monaco, C.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Wong, S.H.; Lam, K.; Lui, R.; Cheung, K.; Tang, W.; Ching, J.Y.; Chan, P.K.; Chan, M.C.; Wu, J.C. Bacteriophage transfer during faecal microbiota transplantation in Clostridium difficile infection is associated with treatment outcome. Gut 2018, 67, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Satoh, N.; Satou, Y.; Davidson, B.; Levine, M. Ciona intestinalis: An emerging model for whole-genome analyses. Trends Genet. 2003, 19, 376–381. [Google Scholar] [CrossRef]
- Dehal, P.; Satou, Y.; Campbell, R.; Chapman, J.; Degnan, B.; De Tomaso, A.; Davidson, B.; Di Gregorio, A.; Gelpke, M.; Goodstein, D.; et al. The draft genome of Ciona intestinalis: Insights into chordate and vertebrate origins. Science 2002, 298, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Leigh, B.A.; Djurhuus, A.; Breitbart, M.; Dishaw, L.J. The gut virome of the protochordate model organism, Ciona intestinalis subtype A. Virus Res. 2018, 244, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.A.; Vaughn, M.; McKay, S.; Lyons, E.; Stapleton, A.E.; Gessler, D.; Matasci, N.; Wang, L.; Hanlon, M.; Lenards, A.; et al. The iPlant collaborative: Cyberinfrastructure for plant biology. Front. Plant Sci. 2011, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Tournayre, J.; Mahul, A.; Debroas, D.; Enault, F. Metavir 2: New tools for viral metagenome comparison and assembled virome analysis. BMC Bioinform. 2014, 15, 76. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Faubladier, M.; Mahul, A.; Paulhe, N.; Bernard, A.; Debroas, D.; Enault, F. Metavir: A web server dedicated to virome analysis. Bioinformatics 2011, 27, 3074–3075. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Cherwa, J.E.; Fane, B.A. Microviridae: Microviruses and Gokushoviruses. In eLS; John Wiley & Sons, Ltd.: Chichester, UK, 2001. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.; Brister, J.R. How to name and classify your phage: An informal guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Labonté, J.M.; Suttle, C.A. Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 2013, 7, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Quaiser, A.; Dufresne, A.; Ballaud, F.; Roux, S.; Zivanovic, Y.; Colombet, J.; Sime-Ngando, T.; Francez, A.-J. Diversity and comparative genomics of Microviridae in Sphagnum-dominated peatlands. Front. Microbiol. 2015, 6, 375. [Google Scholar] [CrossRef] [PubMed]
- Bryson, S.J.; Thurber, A.R.; Correa, A.; Orphan, V.J.; Vega Thurber, R. A novel sister clade to the enterobacteria microviruses (family Microviridae) identified in methane seep sediments. Environ. Microbiol. 2015, 17, 3708–3721. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.; Bawuro, M.; Christopher, A.; Knight, A.; Kraberger, S.; Stainton, D.; Chapman, H.; Varsani, A. Novel single-stranded DNA virus genomes recovered from chimpanzee feces sampled from the Mambilla Plateau in Nigeria. Genome Announc. 2017, 5, e01715–e01716. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Dayaram, A.; Marinov, M.; Ware, J.; Kraberger, S.; Stainton, D.; Breitbart, M.; Varsani, A. Diverse circular ssDNA viruses discovered in dragonflies (Odonata: Epiprocta). J. Gen. Virol. 2012, 93, 2668–2681. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Robin, A.; Ravet, V.; Personnic, S.; Theil, S.; Colombet, J.; Sime-Ngando, T.; Debroas, D. Assessing the diversity and specificity of two freshwater viral communities through metagenomics. PLoS ONE 2012, 7, e33641. [Google Scholar] [CrossRef] [PubMed]
- Kraberger, S.; Waits, K.; Ivan, J.; Newkirk, E.; VandeWoude, S.; Varsani, A. Identification of circular single-stranded DNA viruses in faecal samples of Canada lynx (Lynx canadensis), moose (Alces alces) and snowshoe hare (Lepus americanus) inhabiting the Colorado San Juan Mountains. Infect. Genet. Evol. 2018, 64. [Google Scholar] [CrossRef] [PubMed]
- Shu, P.; Butt, A.M.; Mi, Z.; Wang, W.; An, X.; Pei, G.; Zhang, Z.; Huang, Y.; Zhang, X.; Shi, T. Identification and genomic analysis of a novel member of Microviridae, IME-16, through high-throughput sequencing. Virol. Sin. 2015, 30, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-T.; He, J.-Z.; Zhang, L.-M.; Han, L.-L. Viral metagenomics analysis and eight novel viral genomes identified from the Dushanzi mud volcanic soil in Xinjiang, China. J. Soils Sediments 2018. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart model selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed]
- Stöver, B.C.; Müller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Youens-Clark, K.; Roux, S.; Hurwitz, B.L.; Sullivan, M.B. iVirus: Facilitating new insights in viral ecology with software and community data sets imbedded in a cyberinfrastructure. ISME J. 2017, 11, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P. Vegan: Community Ecology; R Package Version 1.18-28/r1569; 2013. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Kim, K.-H.; Bae, J.-W. Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl. Environ. Microbiol. 2011, 77, 7663–7668. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-H.; Chang, H.-W.; Nam, Y.-D.; Roh, S.W.; Kim, M.-S.; Sung, Y.; Jeon, C.O.; Oh, H.-M.; Bae, J.-W. Amplification of uncultured single-stranded DNA viruses from rice paddy soil. Appl. Environ. Microbiol. 2008, 74, 5975–5985. [Google Scholar] [CrossRef] [PubMed]
- Doore, S.M.; Fane, B.A. The Microviridae: Diversity, assembly, and experimental evolution. Virology 2016, 491, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, M.; Benno, Y. Reclassification of Bacteroides distasonis, Bacteroides goldsteinii and Bacteroides merdae as Parabacteroides distasonis gen. nov., comb. nov., Parabacteroides goldsteinii comb. nov. and Parabacteroides merdae comb. nov. Int. J. Syst. Evol. Microbiol. 2006, 56, 1599–1605. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Semenkovich, N.P.; Whiteson, K.; Rohwer, F.; Gordon, J.I. Going viral: Next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol. 2012, 10, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, D.; Renuka, K. The gut virome: A neglected actor in colon cancer pathogenesis. Futur. Med. 2017, 12, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, T.S.; Xu, Z.; Hansen, M.A.; Sørensen, S.J.; Hansen, L.H. Hundreds of circular novel plasmids and DNA elements identified in a rat cecum metamobilome. PLoS ONE 2014, 9, e87924. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Breitbart, M.; Lee, W.H.; Run, J.-Q.; Wei, C.L.; Soh, S.W.L.; Hibberd, M.L.; Liu, E.T.; Rohwer, F.; Ruan, Y. RNA viral community in human feces: Prevalence of plant pathogenic viruses. PLoS Biol. 2005, 4, e3. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Fornaciari, G.; Luciani, S.; Dowd, S.E.; Toranzos, G.A.; Marota, I.; Cano, R.J. Natural mummification of the human gut preserves bacteriophage DNA. FEMS Microbiol. Lett. 2015, 363, fnv219. [Google Scholar] [CrossRef] [PubMed]
- McCann, A.; Ryan, F.J.; Stockdale, S.R.; Dalmasso, M.; Blake, T.; Ryan, C.A.; Stanton, C.; Mills, S.; Ross, P.R.; Hill, C. Viromes of one year old infants reveal the impact of birth mode on microbiome diversity. PeerJ 2018, 6, e4694. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Nilsson, C.; Lim, Y.W.; Ruan, Y.; Breitbart, M. Metagenomic analysis of viruses in reclaimed water. Environ. Microbiol. 2009, 11, 2806–2820. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.; Kailasan, S.; Cohen, A.; Roux, S.; Tucker, K.P.; Shevenell, A.; Agbandje-McKenna, M.; Breitbart, M. Diversity of environmental single-stranded DNA phages revealed by PCR amplification of the partial major capsid protein. ISME J. 2014, 8, 2093–2103. [Google Scholar] [CrossRef] [PubMed]
- Pearson, V.M.; Caudle, S.B.; Rokyta, D.R. Viral recombination blurs taxonomic lines: Examination of single-stranded DNA viruses in a wastewater treatment plant. PeerJ 2016, 4, e2585. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Guidoni, B.; Jacas, L.; Jacquet, S. Structure and diversity of ssDNA Microviridae viruses in two peri-alpine lakes (Annecy and Bourget, France). Res. Microbiol. 2015, 166, 644–654. [Google Scholar] [CrossRef] [PubMed]
- López-Bueno, A.; Tamames, J.; Velázquez, D.; Moya, A.; Quesada, A.; Alcamí, A. High diversity of the viral community from an Antarctic lake. Science 2009, 326, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Van Bonn, W.; Aw, T.G.; Rose, J.B. Aquarium viromes: Viromes of human-managed aquatic systems. Front. Microbiol. 2017, 8, 1231. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Salamon, P.; Andresen, B.; Mahaffy, J.M.; Segall, A.M.; Mead, D.; Azam, F.; Rohwer, F. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. USA 2002, 99, 14250–14255. [Google Scholar] [CrossRef] [PubMed]
- Angly, F.E.; Felts, B.; Breitbart, M.; Salamon, P.; Edwards, R.A.; Carlson, C.; Chan, A.M.; Haynes, M.; Kelley, S.; Liu, H.; et al. The marine viromes of four oceanic regions. PLoS Biol. 2006, 4, e368. [Google Scholar] [CrossRef] [PubMed]
- Bench, S.R.; Hanson, T.E.; Williamson, K.E.; Ghosh, D.; Radosovich, M.; Wang, K.; Wommack, K.E. Metagenomic characterization of Chesapeake Bay virioplankton. Appl. Environ. Microbiol. 2007, 73, 7629–7641. [Google Scholar] [CrossRef] [PubMed]
- Labonté, J.; Suttle, C. Metagenomic and whole-genome analysis reveals new lineages of gokushoviruses and biogeographic separation in the sea. Front. Microbiol. 2013, 4, 404. [Google Scholar] [CrossRef] [PubMed]
- Labonté, J.M.; Hallam, S.J.; Suttle, C.A. Previously unknown evolutionary groups dominate the ssDNA gokushoviruses in oxic and anoxic waters of a coastal marine environment. Front. Microbiol. 2015, 6, 315. [Google Scholar] [CrossRef] [PubMed]
- Tucker, K.P.; Parsons, R.; Symonds, E.M.; Breitbart, M. Diversity and distribution of single-stranded DNA phages in the North Atlantic Ocean. ISME J. 2011, 5, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-T.; Han, L.-L.; Zhang, L.-M.; He, J.-Z. Diversity and distribution characteristics of viruses in soils of a marine-terrestrial ecotone in East China. Microb. Ecol. 2018, 75, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Mochizuki, T.; Urayama, S.-I.; Yoshida-Takashima, Y.; Nishi, S.; Hirai, M.; Nomaki, H.; Takaki, Y.; Nunoura, T.; Takai, K. Quantitative viral community DNA analysis reveals the dominance of single-stranded DNA viruses in offshore upper bathyal sediment from Tohoku, Japan. Front. Microbiol. 2018, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Desnues, C.; Rodriguez-Brito, B.; Rayhawk, S.; Kelley, S.; Tran, T.; Haynes, M.; Liu, H.; Furlan, M.; Wegley, L.; Chau, B.; et al. Biodiversity and biogeography of phages in modern stromatolites and thrombolites. Nature 2008, 452, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.; Jeffries, T.C.; Roudnew, B.; Seymour, J.R.; Fitch, A.J.; Simons, K.L.; Speck, P.G.; Newton, K.; Brown, M.H.; Mitchell, J.G. Confined aquifers as viral reservoirs. Environ. Microbiol. Rep. 2013, 5, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Takaki, Y.; Eitoku, M.; Nunoura, T.; Takai, K. Metagenomic analysis of viral communities in (hado) pelagic sediments. PLoS ONE 2013, 8, e57271. [Google Scholar] [CrossRef] [PubMed]
- Han, L.-L.; Yu, D.-T.; Zhang, L.-M.; Shen, J.-P.; He, J.-Z. Genetic and functional diversity of ubiquitous DNA viruses in selected Chinese agricultural soils. Sci. Rep. 2017, 7, 45142. [Google Scholar] [CrossRef] [PubMed]
- Reavy, B.; Swanson, M.M.; Cock, P.J.; Dawson, L.; Freitag, T.E.; Singh, B.K.; Torrance, L.; Mushegian, A.R.; Taliansky, M. Distinct circular single-stranded DNA viruses exist in different soil types. Appl. Environ. Microbiol. 2015, 81, 3934–3945. [Google Scholar] [CrossRef] [PubMed]
- Basso, M.F.; da Silva, J.C.F.; Fajardo, T.V.M.; Fontes, E.P.B.; Zerbini, F.M. A novel, highly divergent ssDNA virus identified in Brazil infecting apple, pear and grapevine. Virus Res. 2015, 210, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Neave, M.J.; Apprill, A.; Ferrier-Pagès, C.; Voolstra, C.R. Diversity and function of prevalent symbiotic marine bacteria in the genus Endozoicomonas. Appl. Microbiol. Biotechnol. 2016, 100, 8315–8324. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, L.; Kjeldsen, K.U.; Funch, P.; Jensen, J.; Obst, M.; López-Legentil, S.; Schramm, A. Endozoicomonas are specific, facultative symbionts of sea squirts. Front. Microbiol. 2016, 7, 1042. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Prangishvili, D.; Hendrix, R.W.; Bamford, D.H. Genomics of bacterial and archaeal viruses: Dynamics within the prokaryotic virosphere. Microbiol. Mol. Biol. Rev. 2011, 75, 610–635. [Google Scholar] [CrossRef] [PubMed]
- Leigh, B.A.; Liberti, A.; Dishaw, L.J. Generation of germ-free Ciona intestinalis for studies of gut-microbe interactions. Front. Microbiol. 2016, 7, 2092. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Creasy, A.; Rosario, K.; Leigh, B.A.; Dishaw, L.J.; Breitbart, M. Unprecedented Diversity of ssDNA Phages from the Family Microviridae Detected within the Gut of a Protochordate Model Organism (Ciona robusta). Viruses 2018, 10, 404. https://doi.org/10.3390/v10080404
Creasy A, Rosario K, Leigh BA, Dishaw LJ, Breitbart M. Unprecedented Diversity of ssDNA Phages from the Family Microviridae Detected within the Gut of a Protochordate Model Organism (Ciona robusta). Viruses. 2018; 10(8):404. https://doi.org/10.3390/v10080404
Chicago/Turabian StyleCreasy, Alexandria, Karyna Rosario, Brittany A. Leigh, Larry J. Dishaw, and Mya Breitbart. 2018. "Unprecedented Diversity of ssDNA Phages from the Family Microviridae Detected within the Gut of a Protochordate Model Organism (Ciona robusta)" Viruses 10, no. 8: 404. https://doi.org/10.3390/v10080404
APA StyleCreasy, A., Rosario, K., Leigh, B. A., Dishaw, L. J., & Breitbart, M. (2018). Unprecedented Diversity of ssDNA Phages from the Family Microviridae Detected within the Gut of a Protochordate Model Organism (Ciona robusta). Viruses, 10(8), 404. https://doi.org/10.3390/v10080404